






 本文探讨了如何从SCI高分文章中提取颜色灵感,并在R中进行图表颜色搭配,以增强数据可视化的吸引力。通过使用RImagePalette包读取并提取图片颜色,以及ggsci包提供的预设色彩方案,可以轻松改善图表的视觉效果。此外,作者鼓励读者尝试自己创造色彩搭配,以打造独具个性的图表。
本文探讨了如何从SCI高分文章中提取颜色灵感,并在R中进行图表颜色搭配,以增强数据可视化的吸引力。通过使用RImagePalette包读取并提取图片颜色,以及ggsci包提供的预设色彩方案,可以轻松改善图表的视觉效果。此外,作者鼓励读者尝试自己创造色彩搭配,以打造独具个性的图表。
看SCI文章,发现一个规律,越是高分的文章作图越是“花里胡哨”,出各种新奇的图,色彩上也很鲜艳,而大多数人就喜欢看那种花里胡哨的。其实很多时候,我们里高分文章的图只差一个思路和配色,同样的数据别人做的图看起来很好,很大原因是颜色搭配的好。今天我们就谈谈R的色彩搭配,让你的作图可视化向高分文章看齐。
一、提取颜色
看到别人文章中好看的配色,直接提取是最方便的方法,可以使用AI或者PPT操作,主要是为了得到颜色的十六进制代码,可用于R绘图。不过这里我们介绍一种R的方式,可以将图片读入R中,提取一张图片所有颜色。
首先截取文献的图片,尽量清晰,然后读入
install.packages("RImagePalette")
library(RImagePalette)
library(png)
setwd("F:/生物信息学/图片读入及颜色提取")
picture <- readPNG("1648549584(1).png")
r <- nrow(picture)/ncol(picture)
plot(c(0,1),c(0,r),type = "n",xlab = "",ylab = "",asp=1)
rasterImage(picture,0,0,1,r)
#display_image(picture)
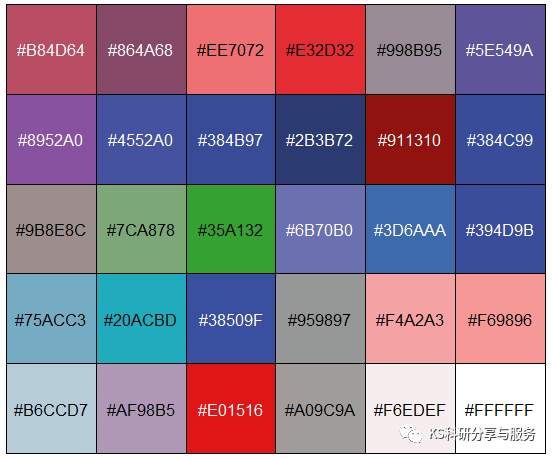
然后尽可能多的获取颜色,并查看颜色,然后自行搭配。
mycolor<-image_palette(picture,n=30)
scales::show_col(mycolor)
mycolor
#"#B84D64" "#864A68" "#EE7072" "#E32D32" "#998B95" "#5E549A" "#8952A0" "#4552A0" "#384B97" "#2B3B72" "#911310"
#"#384C99" "#9B8E8C" "#7CA878" "#35A132" "#6B70B0" "#3D6AAA" "#394D9B" "#75ACC3" "#20ACBD" "#38509F" "#959897"
#"#F4A2A3" "#F69896" "#B6CCD7" "#AF98B5" "#E01516" "#A09C9A" "#F6EDEF" "#FFFFFF"
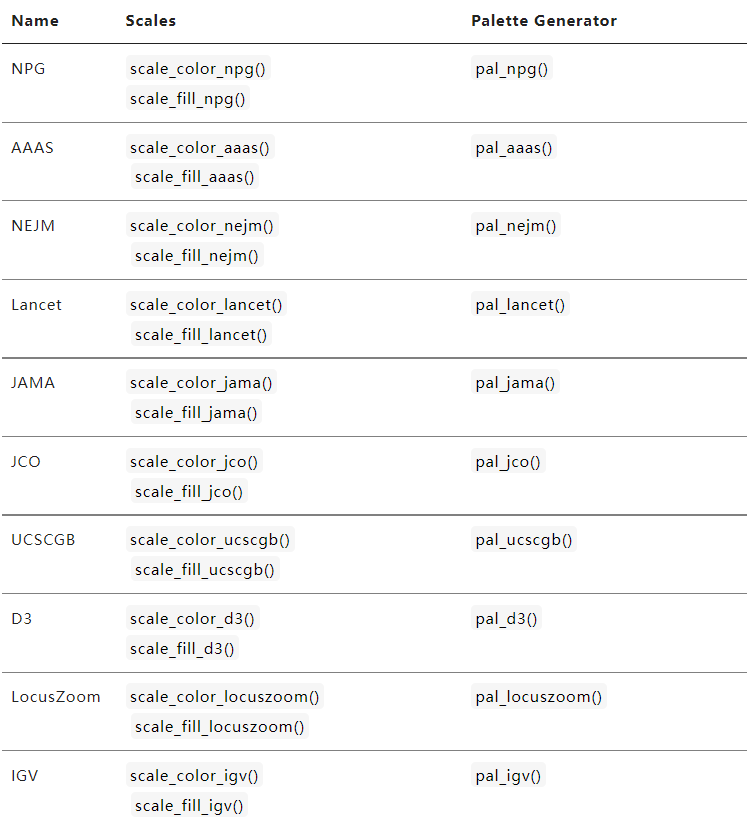
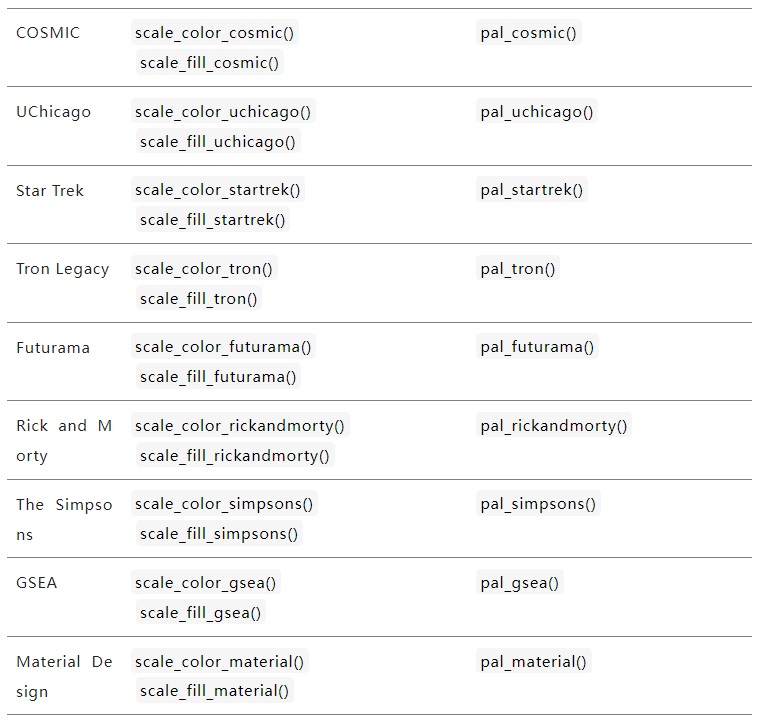
二、自动色彩搭配R包
还记得我们在说单细胞UMAP修饰的时候(单细胞基因可视化之UMAP图修饰),里面提到过一个函数ggsci。这个包提供了各种形式的色彩搭配,有适用于连续变量的--=例如热图,也有离散性变量的颜色搭配,还是比较方便的。与ggplot2配合使用,做好图之后,选择需要的主题修饰即可。
library(ggsci)
p
p+scale_color_npg()
以下是ggsci网站提供的各种主题函数汇总:https://nanx.me/ggsci/articles/ggsci.html
三、自己造
要说最简单粗暴、最有个性的搭配还是靠自己,这里分享R语言颜色表以供参考,还请不要太过“浓妆艳抹”。想要获取完整版的可以在《KS科研分享与服务》公众号后台回复:R语言颜色,自行下载。
快去修改下自己图的颜色吧,让其变个装!

















 1784
1784

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









