







 介绍MEGAHIT基因组组装工具的原理、安装、使用方法及常见问题解决。适用于处理宏基因组数据,采用迭代kmer的de Bruijn Graph算法实现快速高效的组装。
介绍MEGAHIT基因组组装工具的原理、安装、使用方法及常见问题解决。适用于处理宏基因组数据,采用迭代kmer的de Bruijn Graph算法实现快速高效的组装。
简介
宏基因组测序获得海量短片段测序数据,这些数据混合着环境中各种各样的微生物基因组序列,如何恢复出这些微生物基因组序列,基因组组装成为至关重要的一步。
在考虑如此复杂的数据之前,不妨先看看单个基因组组装的常规步骤:
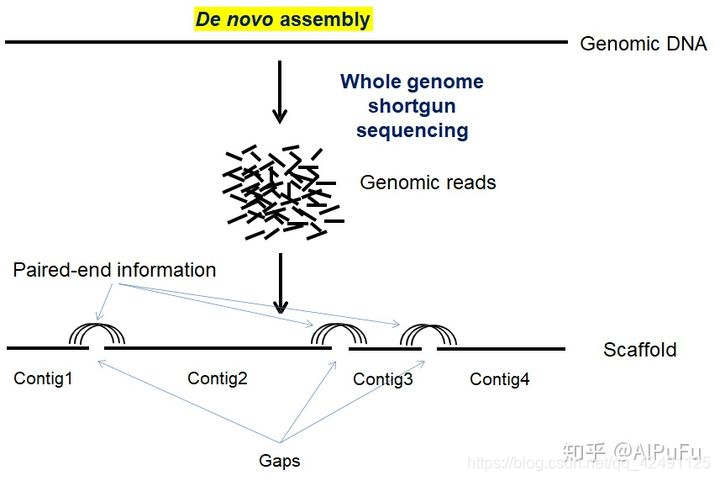
- 首先,通过shotgun测序产生reads,然后利用连续reads之间的重叠信息(overlap)进行组装产生长片段Contig
- 其次,可以使用pair-end之间的位置信息确定Contig的方向和顺序,组装产生更长的片段Scaffold,
- 最后,可以基于大片段的文库组装连接Scaffold得到完整的染色体序列
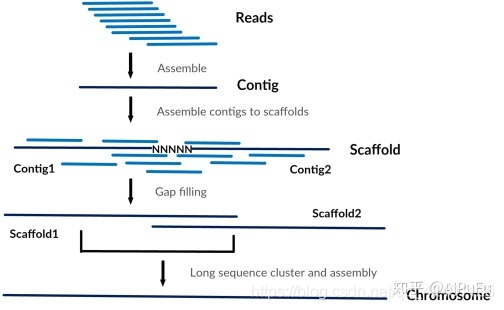
需要说明的是,之所以可以使用pair-end的信息连接Contg,是因为一般在建库的时候长度要大于实际测序的总长度,例如建库常用500bp,而Illumina pair-end 150bp加到一起才300bp,因此中间200bp的位置信息在第一步组装成Contig时实际是没有利用到的。
组装算法大致可以分为三类: - 1.基于OLC(Overlap-Layout-Consensus),适用于测序量不大的长片短数据
- 2.基于kmer的DBG(de bruijn graph),适用测序量大的段片段数据
- 3.综合OLC和DBG法
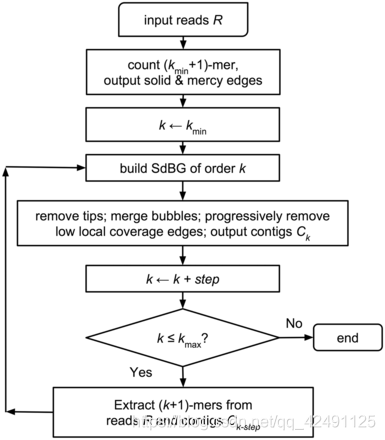
MEGAHIT采取的算法则是基于迭代的kmer的DBG法,其特点是超快和超高效内存使用
-
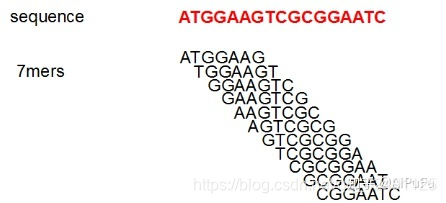
1)把所有测序读段(reads)都分割为更小的片段k-mer:这里k为7,假如read的长度为n,则总共可产生n-k+1个k-mer;
-
2)把每个k-mer作为一个节点,然后判断k-mer之间是否有k-1碱基的重叠,如果有则将两个不同的节点连接起来。依次这样连接所有可连接的k-mer就形成了de Bruijn Graph;
-
3)依次合并相邻的k-mer,因为相邻的k-mer有k-1个碱基的重叠,就可进一步简化de Bruijn Graph形成简化后的图;
由于相邻k-mer之间的非配对碱基只有1个,那理论上相邻序列的组合情况只有四种,这大大减少了相邻序列配对的检测难度,因此de Bruijn方法是现阶段的主流组装方法。 -
4)使用一系列算法消除由测序错误而形成的tips,并合并bubbles(两条或多条路径序列,一般由SNP或者测序错误造成;
-
5)增加kmer长度,重新执行上述步骤,如果结果不好于上一步则终止迭代,否则继续迭代。

MEGAHIT paperIF: 5.8 Q1 B3
安装和使用
# 1.Conda安装
conda install -c bioconda megahit
# 2. 查看安装版本
megahit –v
#MEGAHIT v1.2.9
- 输入文件:fastq/fasta格式的测序文件(单端或双端),可以是gz/bz2压缩文件
以常见的fastq格式文件为例:
- 使用
# 双端序列组装:
megahit -1 pe_1.fq -2 pe_2.fq -o out
#-1:pair-end 1序列,-2 pair-end 2序列,-o输出目录
# 单端序列:
megahit -r single_end.fq -o out
# 交错的双端序列:
megahit --12 interleaved.fq -o out
- 常用参数
两种设置kmer参数的方法:
1)–k-list:组装的kmer size列表,支持多kmer组装,不同kmer size之间逗号分隔,可设置的范围15-255,相邻kmer size间隔必须小于或等于28,默认为21,29,39,59,79,99,119,141
2)组合参数:–k-min:设置最小的kmer size,应小于255,必须为奇数,默认为21
–k-max:设置最大的kmer size,应小于255,必须为奇数,默认为141
–k-step:多kmer组装的kmer size间隔,应小于等于28必须为偶数,默认为12
示例:
megahit -r single_end.fq -o out --k-list 21,39,59
megahit -r single_end.fq -o out --k-min 21 --k-max 141 --k-step 10
- 其他常用参数
1)-m/–memory:构建SdBG可以使用的最大内存,可设置0-1,也即占总内存的分数,默认为0.9
2)-t/–num-cpu-threads:程序运行使用的核数
3)–out-prefix:输出结果文件的前缀,例如contig文件会是OUT_DIR/OUT_PREFIX.contigs.fa
4)–min-contig-len输出的最短contigs,默认为200
5)–test 在自带测试数据上执行组装,常用于测试软件安装是否正常
示例:
megahit -r single_end.fq -o out –m 0.5 –t 10
megahit --test
常见报错和解决方法
std::bad_alloc/Exit code -6

解决方法:
1)指定大内存:-m 0.9
2)尝试使用大kmer组装,降低组装复杂度:例如设置–k-list 39,49,…,129,141
3)若上述都无效则需要加大服务器内存
从中断点继续运行
当组装大数据的时候可能由于内存不够或其他原因中断,此使可以使用
megahit --continue -o former_megahit_out -m 0.9
参数说明:
–continue:从中断处继续运行
-o former_megahit_out: 输出目录,必须是之前已经生成的目录,包含相关中间文件
-m 0.9:使用内存百分比,也可加其他参数
输出结果
MEGAHIT输出结果很简单,最重要的就是组装基因组final.contigs.fa文件

final.contigs.fa: 组装结果,fasta格式
log: megahit程序运行时的log日志,便于查看具体执行进度和排错
options.json: 执行组装程序参数的json格式
intermediate_contigs: 中间组装结果
预设参数
megahit有两组预设参数meta-sensitive及meta-large,前者更加灵敏,所耗时间较长;后者比较适合大型且复杂的宏基因组数据,例如土壤。
Presets parameters:
--presets <str> override a group of parameters; possible values:
meta-sensitive: '--min-count 1 --k-list 21,29,39,49,...,129,141'
meta-large: '--k-min 27 --k-max 127 --k-step 10'
(large & complex metagenomes, like soil)
对内存需求
- MEGAHIT的内存需求至少需要原始数据的1.04倍~1.5倍,内存越大越好

内存消耗的峰值在于kmer计数和构建debruijn graph步骤,会将所有reads/edges的读到内存用于排序

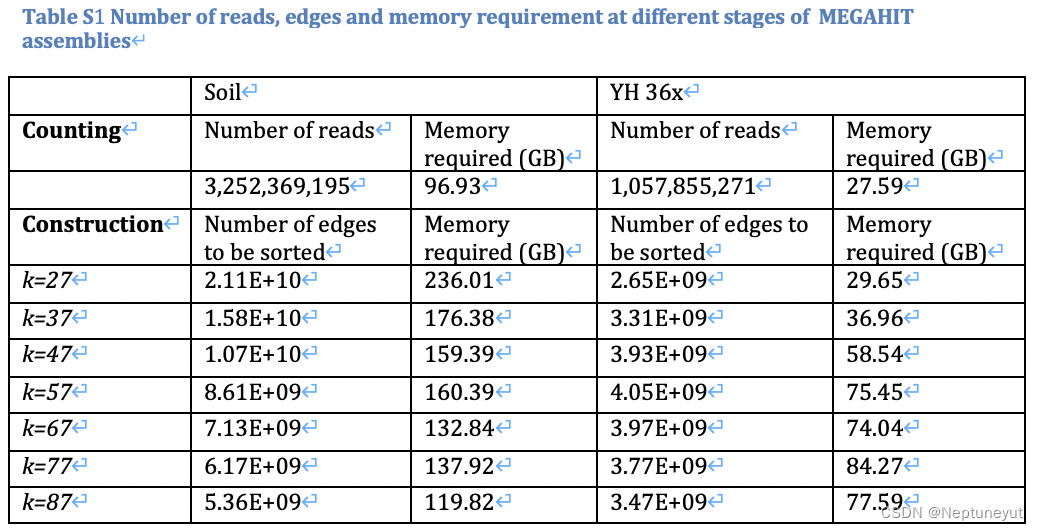
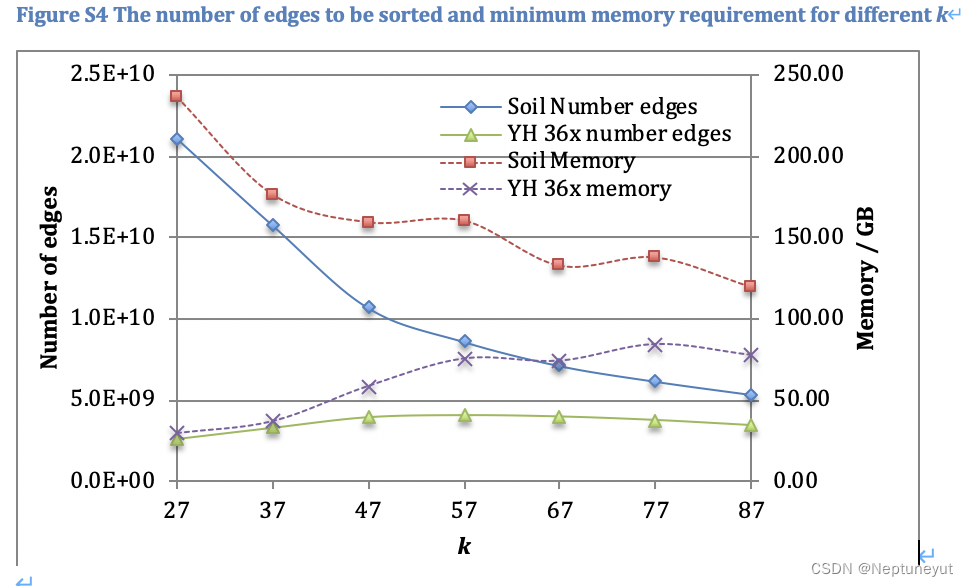
样本实际组装时间
对于双端测序fastq文件,使用30核,每Gb(PE fq文件大小算单端)平均耗时0.35h


















 6756
6756

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









