







小罗碎碎念
推文主题——人工智能在肿瘤细胞分类中的应用。

重点关注
临床方向的同学/老师建议重点关注第四篇&第六篇文章,最近DNA甲基化和蛋白组学与AI的结合,在顶刊中出现的频率很高,建议思考一下能否和自己的课题结合。
工科的同学重点所有的文章都值得关注,毕竟小罗就是从工科角度出发挑的这些文章,其中第五篇是一定要看的,后期我把病理组学的文章写完就来分析这篇。
如果您的课题与病理组学相关,强烈推荐第二篇文章,可以打开你的思路。另外,小罗感觉自己之前对于多组学的理解有点片面了,只停留在最基础的几种组学,没有考虑到代谢组学和蛋白组学等,希望你们看完这篇推文也能有所启发。
一、AIEgen荧光探针,结合分割一切模型精确识别癌细胞

文献概述
这篇文章是关于一种名为
AIEgen-Deep的创新分类程序的研究,该程序结合了AIEgen荧光染料、深度学习算法和Segment Anything Model(SAM),用于精确识别癌细胞。
AIEgen-Deep通过显著减少80%-90%的手动注释工作量,展示了在识别癌细胞形态方面的显著准确性,在26,693张八种不同细胞类型的图像上达到了75.9%的准确率。
在健康细胞与癌细胞的二元分类中,该模型显示出增强的性能,准确率为88.3%,召回率为79.9%。该模型能有效区分健康细胞(成纤维细胞和白细胞)和不同类型的癌细胞(乳腺癌、膀胱癌和间皮瘤细胞),准确率分别为89.0%、88.6%和83.1%。预计该方法在不同癌症类型的广泛应用将显著有助于早期癌症检测并提高患者的生存率。
知识点补充:
准确率VS召回率
准确率(Accuracy)和召回率(Recall),也称为真正例率(True Positive Rate),是评估分类模型性能的两个重要指标,尤其在二分类问题中经常使用。
准确率(Accuracy):
- 准确率是分类模型正确预测的样本数占总样本数的比例。
- 它通过将
正确预测的正例(真正例)和负例(真负例)的数量相加,然后除以总样本数来计算。 - 公式为:
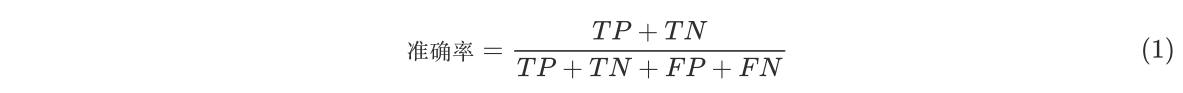
- 其中,TP(真正例)是模型正确预测为正的样本数,TN(真负例)是模型正确预测为负的样本数,FP(假正例)是模型错误预测为正的样本数,FN(假负例)是模型错误预测为负的样本数。
- 准确率提供了模型整体性能的快照,但在不平衡的数据集中可能不是最佳性能指标,因为它可能会受到多数类的过度影响。
召回率(Recall):
- 召回率衡量的是模型正确识别为正的样本占所有实际正样本的比例。
- 它只关注正类样本,即它计算的是所有实际为正的样本中,模型正确预测为正的比例。
- 公式为:
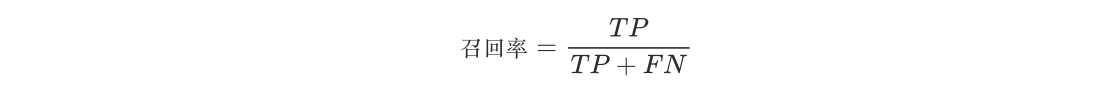
- 召回率对于评估模型检测所有重要事件的能力非常重要,尤其是在那些漏检(未被模型检测到的正样本)的代价很高的应用中,如疾病筛查、欺诈检测等。
在实际应用中,准确率和召回率之间往往存在权衡关系。一个高准确率的模型可能在少数类上表现不佳,导致召回率较低;而一个高召回率的模型可能会增加误报(将负样本错误预测为正样本),从而降低准确率。因此,选择哪个指标作为优化目标,需要根据具体问题和业务需求来确定。在某些情况下,可能需要使用其他指标如F1分数或接收者操作特征曲线(ROC曲线)下的面积(AUC)来进行更全面的模型评估。
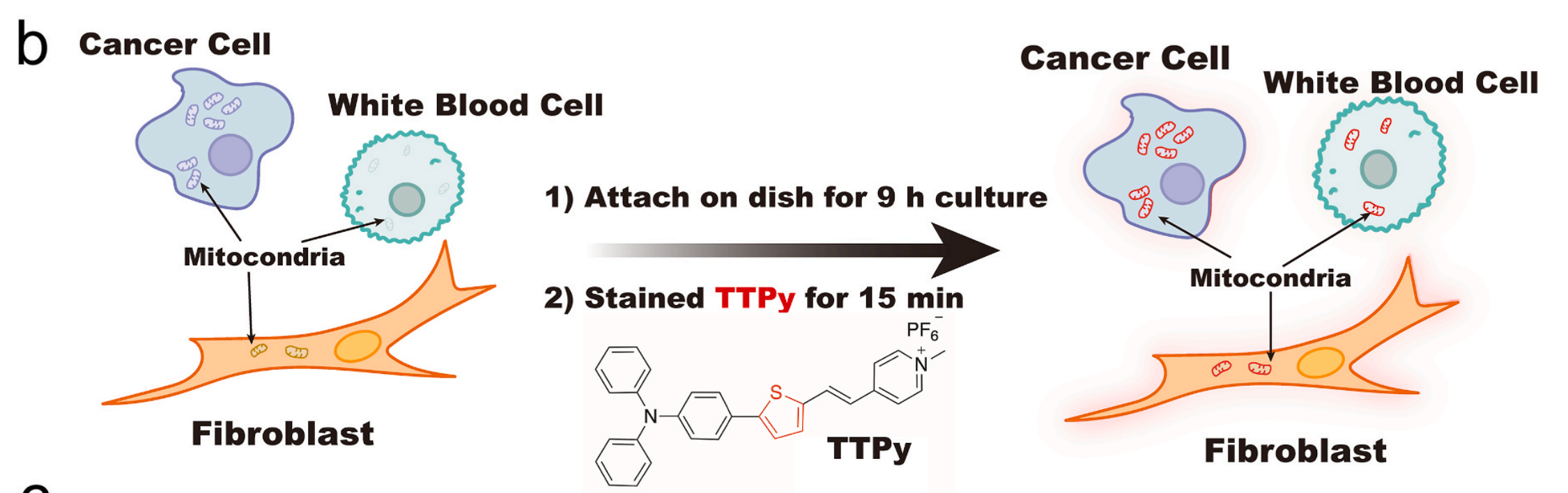
研究还介绍了AIEgen-Deep的开发过程,包括设计和合成了一种名为TTPy的近红外发射AIEgen分子,专门用于
线粒体靶向。
TTPy分子具有固有的带正电荷的基团,使其适合靶向线粒体。该分子通过Suzuki和Knoevenagel反应合成,并研究了其光物理性质和生物成像能力。
研究结果表明,TTPy在480nm处有最大吸收,在670nm处有最大发射,Stokes位移显著,有利于避免生物成像中的“内滤器”效应。此外,TTPy在DMSO/水混合物中的发射强度随水组成的逐渐增加而增强,显示出典型的AIE现象。
知识点补充:线粒体靶向
线粒体靶向(Mitochondria Targeting)是一种将分子、化合物或药物直接传递到细胞内线粒体的技术或方法。线粒体是细胞的能量工厂,它们在细胞代谢、细胞凋亡(程序性死亡)、钙信号传递以及许多其他关键细胞功能中发挥着重要作用。因此,线粒体靶向在药物递送、疾病治疗和生物医学研究中具有重要意义。
线粒体靶向的主要目的包括:
-
提高疗效:通过将药物直接传递到线粒体,可以增加药物在作用部位的浓度,从而提高治疗效果。
-
减少副作用:靶向递送可以减少药物对非靶组织的分布,从而减少副作用和毒性。
-
研究线粒体功能:通过使用线粒体靶向分子,研究人员可以更精确地研究线粒体在各种生物学过程和疾病中的作用。
-
疾病治疗:许多疾病,包括
神经退行性疾病、心血管疾病和某些癌症,与线粒体功能障碍有关。线粒体靶向可用于开发针对这些疾病的新疗法。
实现线粒体靶向的策略通常包括:
-
使用线粒体定位序列:这些是一类短肽,可以与线粒体的外膜特异性结合,用于将药物或分子传递到线粒体。
-
利用线粒体靶向分子:例如,使用带正电的分子或特定化学结构的分子,这些分子可以与线粒体膜的负电荷相互作用。
-
物理方法:如微射流、电穿孔等技术,可以直接将分子送入细胞。
-
化学修饰:通过在药物分子上附加特定的化学基团,可以增强其与线粒体膜的亲和力。
-
纳米技术:利用纳米粒子作为载体,实现对线粒体的靶向药物递送。
在上文提到的AIEgen-Deep研究中,TTPy作为一种近红外发射的聚集诱导发光(AIE)探针,被设计用于靶向线粒体成像。这种探针的开发,使得研究人员能够更精确地观察线粒体的结构和功能,对于癌症等疾病的早期诊断和研究具有重要的应用潜力。
文章还讨论了AIEgen-Deep系统的未来改进,包括在不同环境条件下的广泛测试,以确定其在不同临床和研究成像场景中的适应性和鲁棒性。
最后,研究成功开发了一种新型的近红外发射聚集诱导发光(AIE)探针TTPy,用于靶向线粒体成像。TTPy的独特分子结构,包括带正电荷的基团和分子旋转机制,被合成以增强聚集状态下的发射强度,解决了生物成像中的关键需求。
研究展示了TTPy在包括各种癌细胞和非癌细胞在内的不同细胞类型中优越的线粒体染色效率和光稳定性。
结合使用OpenCV的自动细胞标记算法和AIEgen-Deep深度学习框架的应用,标志着细胞成像的重要进展。
使用先进的算法,如ResNet和Vision Transformer,以及YOLOv5和Faster RCNN,显著提高了异质样本中细胞类型区分和癌细胞检测的准确性和效率。特别是结合AIE探针和Vision Transformer的使用,达到了80.8%的癌症类型分类准确率和75.9%的八种不同细胞类别的准确率。
这项研究使用的26,693张高质量细胞图像的大型数据集,为有效训练深度学习算法提供了坚实可靠的基础,进一步增强了研究结果的鲁棒性和准确性。将人工智能(AI)整合到生物医学中,是一个特别有希望的发展。AI在快速处理和解释大型数据集方面的能力,可以进行更准确和个性化的医学诊断。
AI算法擅长在医学图像中识别复杂模式,可能识别出人类检测不到的异常。这种能力对于像癌症这样的疾病至关重要,早期发现显著影响治疗结果。
在分子成像中,AIE探针的独特特性,如在聚集状态下荧光增强,为高灵敏度和特异性成像方法开辟了新途径。通过将AI分析与这些探针结合,我们可以迅速解释复杂的成像数据,增强我们对细胞动态和疾病演变的理解。
重点关注
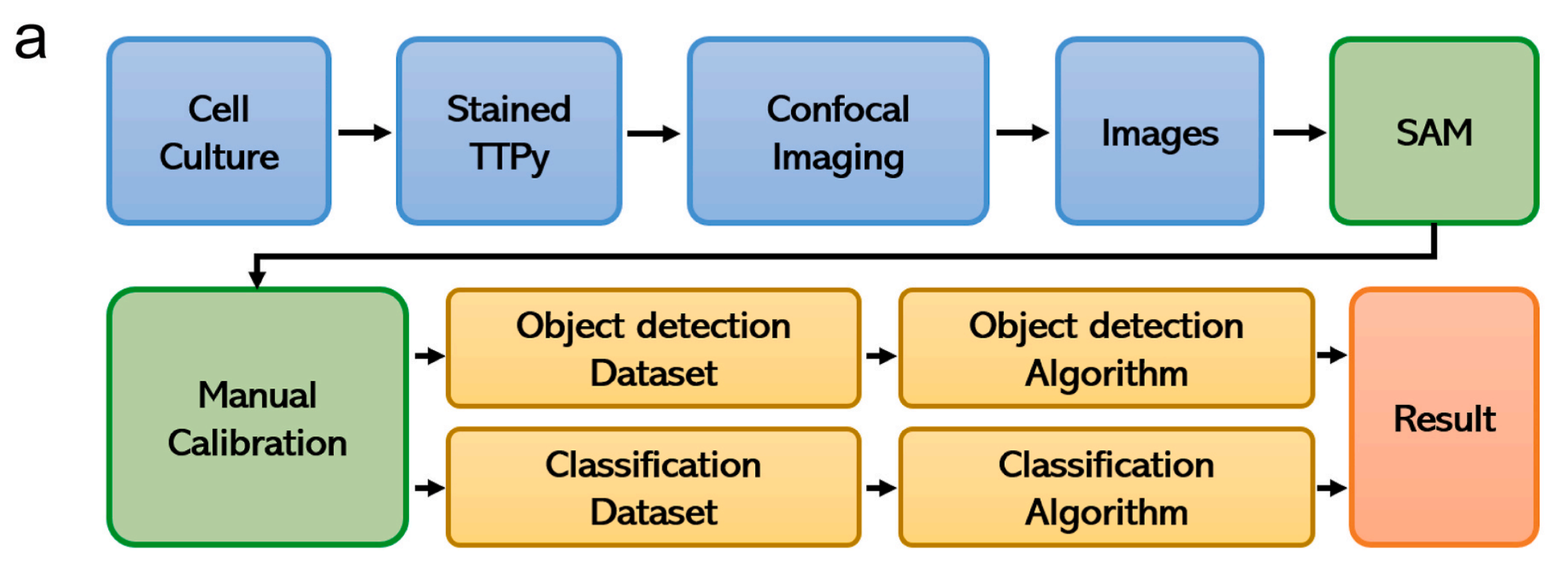
Fig. 2 在文章中提供了AIEgen-Deep学习系统的全面概览以及它在细胞类型识别和分类中的应用。
以下是对图中各部分的分析:
a) 架构概览:
- AIEgen-Deep学习系统的架构从图像处理开始,图像首先通过自动标记算法进行处理。
- 处理后的图像数据被分割成两个数据集:数据集I和数据集II。
- 在数据增强之后,这些数据集被送入深度学习分类和目标识别模型进行进一步分析。
b) 细胞类型的差异化染色:
- AIEgen染料通过不同的染色方式来区分不同类型的细胞,这使得系统能够通过突出显示独特的形态特征来准确识别和分类细胞。
- 这种差异化染色策略是提高细胞识别准确性的关键,因为它允许深度学习模型捕捉到每种细胞类型特有的视觉信息。
c) SAM和ViT架构的详细说明:
- 详细展示了Segment Anything Model (SAM)和Vision Transformer (ViT)这两种先进的深度学习模型的架构和设计组件。
- SAM是一个能够处理图像分割任务的模型,它可以识别图像中的不同对象并进行分类。
- ViT是一种基于Transformer架构的视觉模型,它利用注意力机制来处理图像数据,从而实现高效的图像分析。
- 这两种模型在AIEgen-Deep系统中被用来进行癌症细胞的识别和分类,它们通过分析经过AIEgen染料染色的细胞图像,来区分不同类型的细胞。
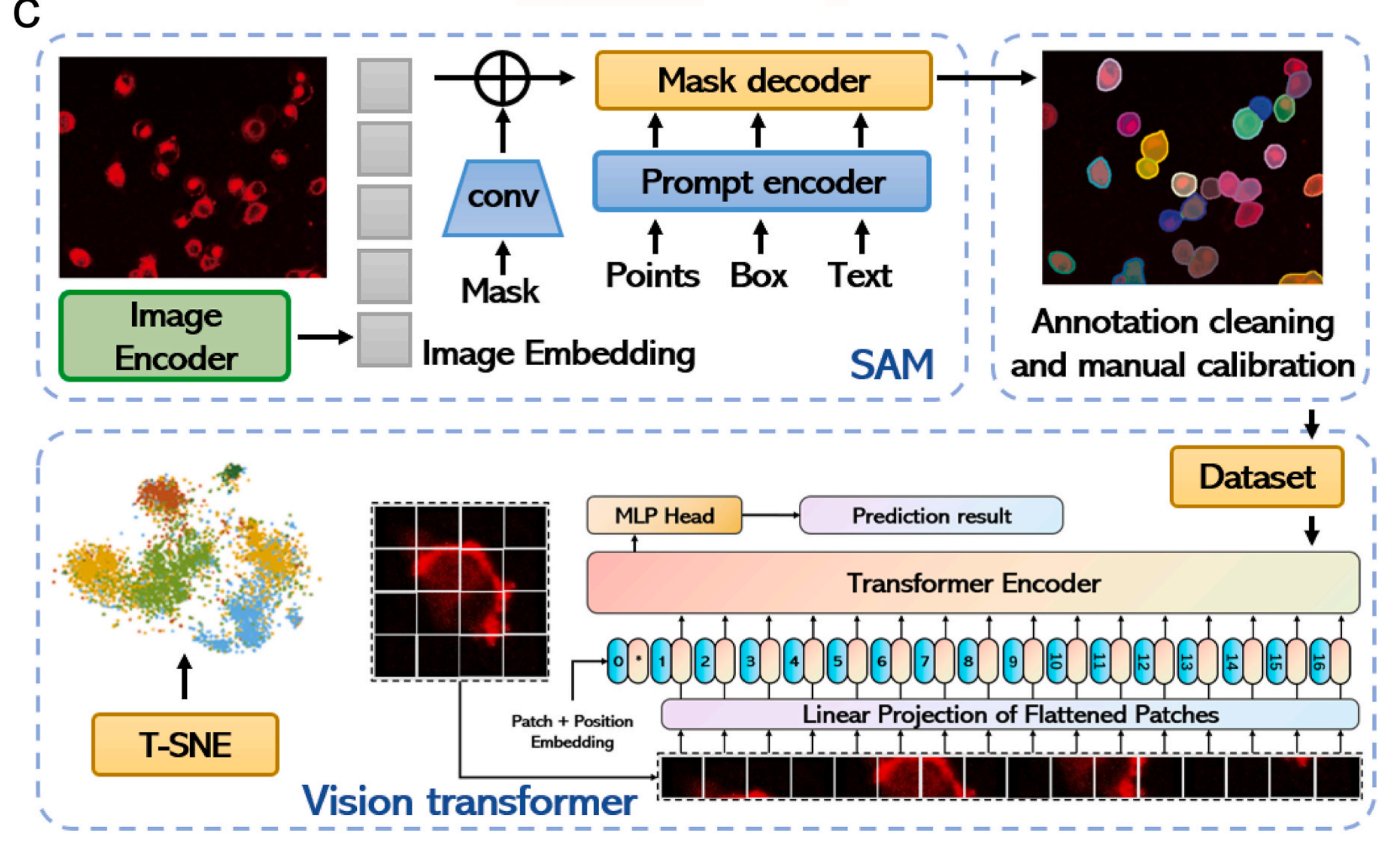
整体而言,Fig. 2 强调了AIEgen-Deep系统如何通过结合先进的成像技术和深度学习算法来实现对复杂生物样本中细胞类型的高效和准确分类。通过自动化的图像处理和数据增强,系统能够训练深度学习模型以识别和分类具有不同形态特征的细胞,这对于癌症的早期诊断和治疗具有重要意义。
二、从WSI级别到patch级别,AI实现双核细胞的细粒度分类

文献概述
这篇文章是关于一种新的医学图像分析方法,旨在提高对
双核细胞(Binuclear Cells, BCs)的检测精度和速度。
BCs的检测对于预测白血病和其他恶性肿瘤的风险具有重要意义。然而,传统的显微镜图像手动计数BCs不仅耗时而且主观性强。此外,由于染色质量和BC显微镜全切片图像(Whole-Slide Images, WSIs)中形态特征的多样性,传统的图像处理方法表现不佳。
知识点补充:双核细胞
双核细胞(Binuclear Cells,简称BCs)是一种具有两个细胞核的异常细胞。在正常情况下,哺乳动物的体细胞通常是单核的,即每个细胞内只有一个细胞核。然而,在某些病理条件或受到某些刺激(如辐射、化学物质、病毒感染等)时,细胞可能会出现异常的细胞分裂,导致形成双核或多核细胞。
双核细胞的形成机制可能包括:
- 细胞分裂失败:在细胞分裂过程中,如果细胞分裂不完全,两个细胞核可能被困在同一个细胞膜内,形成双核细胞。
- 细胞融合:两个相邻的细胞可能因为某些原因融合在一起,导致两个细胞核共存于一个细胞中。
- 异常有丝分裂:在有丝分裂过程中,染色体分配不均或纺锤体功能异常,也可能导致双核细胞的形成。
双核细胞在医学和生物学研究中具有重要意义,因为它们可以作为:
- 遗传毒性测试的指标:在遗传毒性测试中,双核细胞的形成常常被用来评估化学物质或环境因素对DNA的损伤。
- 癌变风险的生物标志物:双核细胞的出现可能与早期肿瘤的发生有关,因此在癌症风险评估中具有指示作用。
- 细胞生物学研究的对象:研究双核细胞有助于我们理解细胞分裂和细胞周期调控的正常和异常机制。
在临床诊断和医学研究中,检测和分析双核细胞对于评估个体的健康状况和疾病风险具有重要价值。
为了克服这些挑战,文章提出了一种基于深度学习的多任务方法。该方法受BC结构先验的启发,分两个阶段实现:首先是在WSI级别进行BC的粗糙检测,然后在补丁(patch)级别进行细粒度分类。
粗糙检测网络
粗糙检测网络是一个基于圆形边界框的多任务检测框架,用于细胞检测和核中心关键点检测。
与常规矩形框相比,圆形表示降低了自由度,减轻了周围杂质的影响,并且可以在WSIs中实现旋转不变性。检测核中的关键点可以帮助网络感知,并用于后续细粒度分类中的无监督颜色层分割。
细粒度分类网络
细粒度分类网络由基于颜色层掩模监督的背景区域抑制模块和基于Transformer的全局建模能力的键区域选择模块组成。
此外,文章首次提出了一个无监督且未配对的细胞质生成器网络,用于扩展长尾分布数据集。
知识点补充:长尾分布
在机器学习和数据科学中,长尾分布数据集指的是某些类别的样本数量远多于其他类别的数据集。例如,在自然语言处理中,常见词(如“the”,“is”)的出现频率可能远远高于一些罕见词的出现频率。在图像识别中,某些类别的图像(如猫和狗)可能有很多样本,而其他类别(如特定种类的植物或罕见动物)的样本则相对较少。
长尾分布数据集带来的挑战包括:
- 类别不平衡:少数类别的样本数量远多于其他类别,导致模型可能偏向于预测那些频繁出现的类别。
- 泛化能力差:如果模型只在大量样本的类别上训练得很好,它可能无法很好地泛化到样本量较少的类别。
- 评估困难:传统的评估指标可能无法准确反映模型在所有类别上的性能,特别是对于那些样本量少的类别。
为了解决长尾分布数据集中的这些问题,研究人员可能会采用以下一些策略:
- 过采样:增加少数类别的样本数量,使其与多数类别的样本数量相匹配。
- 欠采样:减少多数类别的样本数量,以减少其对模型训练的影响。
- 数据增强:对少数类别的样本进行变换,生成更多的样本。
- 集成学习:结合多个模型的预测,以提高对少数类别的识别能力。
- 重采样:在训练过程中动态调整样本的选择,以平衡不同类别的表示。
最终,在BC多中心数据集上进行的实验表明,所提出的BC细粒度检测方法在几乎所有评估标准上都优于其他基准,为癌症筛查等任务提供了明确和支持。
重点关注
Fig. 2 展示了所提出的双核细胞(BC)检测网络的概览。该网络旨在从用Giemsa染色的显微镜全切片图像(WSIs)中提取BC图像数据。
整个流程分为两个主要部分:粗糙检测和细粒度分类。
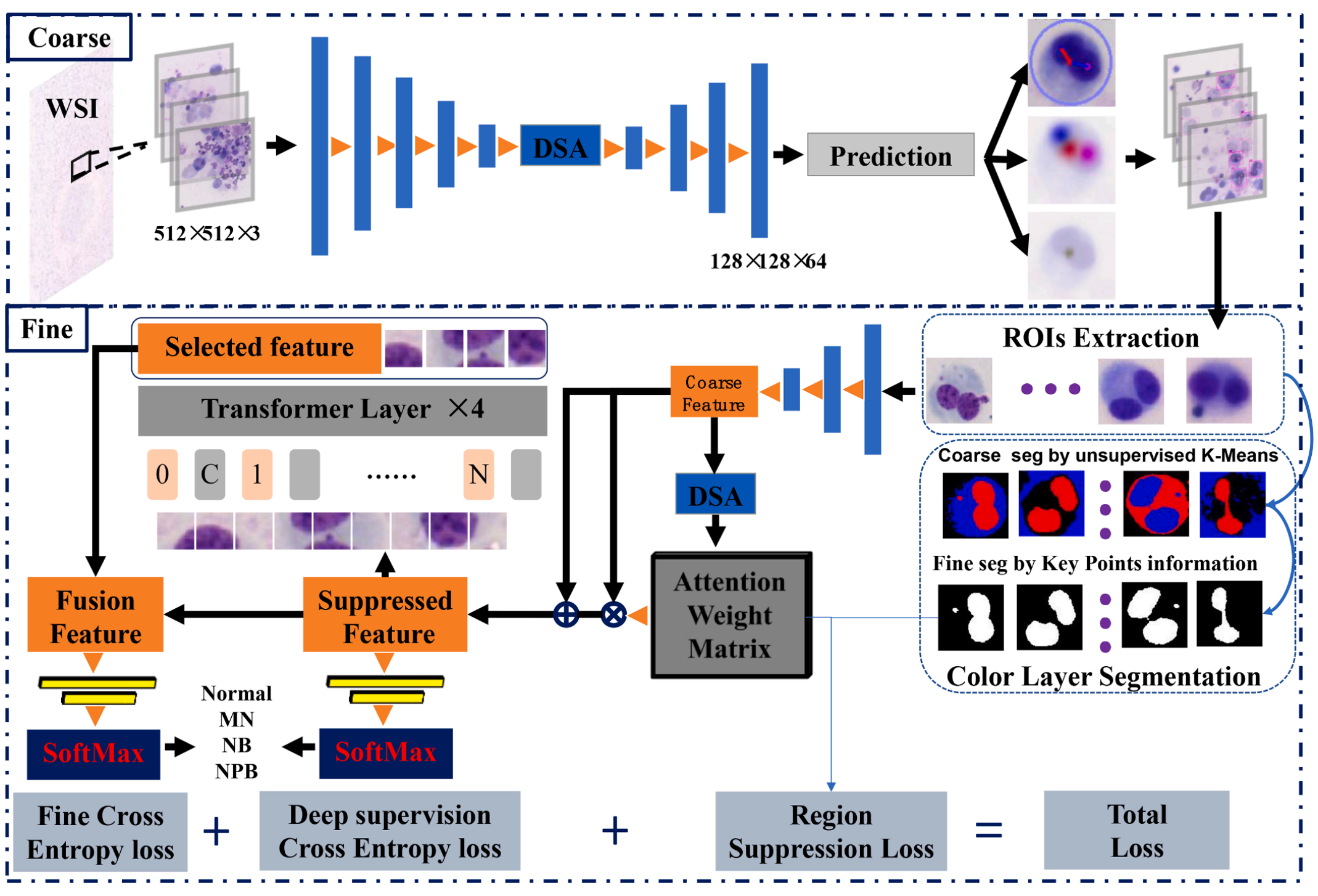
-
粗糙BC检测:在第一部分中,首先进行BC的粗糙检测。这一步骤的目的是识别出BC的大致位置和形状,但不涉及详细的分类。检测到的BC被提取为粗糙感兴趣区域(ROIs)。
-
细粒度分类前的处理:在过渡到细粒度分类阶段之前,文章中提到使用无监督的K-means聚类算法对细胞颜色层进行分割。这一步骤是为了区分图像中的不同区域,如未染色区域、细胞质区域和细胞核区域。通过颜色层分割,可以创建一个掩模(mask),该掩模随后用于细粒度分类中的区域抑制模块。
-
区域抑制模块:在细粒度分类网络中,利用从颜色层分割得到的掩模作为监督特征,对注意力特征图(attention feature map)上的背景区域进行抑制。这意味着网络将减少对背景的关注度,而更多地集中于细胞核等关键区域。
-
基于Transformer的区域选择模块:为了进一步提升分类精度,网络还加入了基于Transformer的区域选择模块。Transformer是一种强大的模型,能够捕捉全局上下文信息,有助于识别图像中的关键特征区域。
-
特征融合与最终分类:最后,经过区域抑制和区域选择处理的特征被融合起来,用于最终的BC分类。这种融合有助于结合局部和全局信息,提高分类的准确性。
总体而言,Fig. 2 描述了一个从粗糙检测到细粒度分类的多阶段处理流程,每个阶段都利用了特定的图像处理技术和深度学习模块,以提高对BC的检测和分类精度。通过这种结构化的方法,网络能够更好地理解和分类BC的不同类型,这对于诸如癌症筛查等应用至关重要。
三、基于Vision Transformer的新型深度学习架构CellViT,实现精确的细胞分割和分类

文献概述
这篇文章是关于一种名为CellViT的新型深度学习架构,它基于Vision Transformer,用于精确的细胞分割和分类。这项研究由德国埃森大学医院(University Hospital Essen)的人工智能医学研究所(Institute for AI in Medicine, IKIM)和癌症研究中心(Cancer Research Center, CCCE)等多个机构的研究人员共同完成。
背景与动机:
在临床任务中,对细胞核的检测和分割在血液和组织样本的分析中至关重要。然而,由于染色和大小的差异、边界重叠以及细胞聚集,这一直是一个挑战性的任务。尽管卷积神经网络(CNNs)已被广泛用于此任务,但作者探索了基于Transformer的网络结合大规模预训练在此领域的潜力。
CellViT架构:
CellViT是一种用于自动化细胞核实例分割的深度学习架构,它使用基于Vision Transformer的学习架构。
CellViT在PanNuke数据集上进行了训练和评估,这是一个包含19种组织类型中近200,000个注释细胞核的、具有挑战性的细胞实例分割数据集。
知识点补充:实例分割
实例分割(Instance Segmentation)是计算机视觉领域中的一项任务,它旨在识别图像中所有感兴趣的实例(即个体对象),并精确地分割它们,同时对每个实例进行分类。这与仅仅识别图像中的类别(分类任务)或将图像分割成不同的区域(语义分割任务)不同,实例分割需要同时完成对象检测、像素级分割和分类。
实例分割通常包括以下几个步骤:
-
对象检测:首先确定图像中所有感兴趣对象的位置,通常以矩形框(bounding box)的形式表示。
-
实例分割:然后对每个检测到的对象实例进行像素级的分割,即使对象属于同一类别,分割也要区分不同的个体实例。
-
分类:最后对每个分割出的实例进行分类,确定它们属于哪个类别。
实例分割在多个应用领域中非常重要,例如自动驾驶车辆中识别和分割行人、车辆等不同实例,或者在医学图像分析中识别和分割不同类型的细胞。
实例分割的挑战在于需要精确的定位和区分图像中所有对象的实例,这通常涉及到复杂的算法和深度学习模型。一些流行的实例分割算法包括Mask R-CNN、YOLACT、TensorMask等,这些模型通常结合了卷积神经网络(CNN)和区域建议网络(RPN)来实现高效的实例分割。
主要贡献:
- 提出了一种新颖的基于U-Net编码器-解码器网络的细胞实例分割方法,利用Vision Transformers作为编码器网络。该方法在PanNuke数据集上实现了优于现有方法的细胞核检测性能,并展示了CellViT在没有微调的情况下应用于MoNuSeg数据集的泛化能力。
- 提供了一个框架,通过使用1024×1024像素的大推理补丁大小,与常规的256像素大小补丁相比,能够快速应用于千兆像素级别的全切片图像(WSI)的推理结果。
- 新架构能够提取与ViT最后一层Transformer中的检测到的细胞对应的细胞嵌入向量。在CoNSeP数据集上的实验表明,这些嵌入向量对于将模型适应新类别非常有用,实现了0.963的线性分类AUROC。
结论:
CellViT证明了大规模领域内和领域外预训练的Vision Transformers的优越性,并展示了在PanNuke数据集上实现最先进的细胞核检测和实例分割性能。
此外,文章还提到了未来的研究方向,包括将提取的细胞嵌入向量应用于下游组织分析任务,以及评估哪种嵌入向量更适合下游任务。
重点关注
CellViT(Vision Transformers for precise cell segmentation and classification)的网络结构。
CellViT 是一种用于精确分割和分类细胞核的深度学习模型,其网络结构具有以下几个关键特点:

-
输入图像转换为Token序列:输入的图像首先被转换成一系列的token(即图像的展平输入部分)。这些token是图像的局部区域,它们将作为Transformer模型的基本输入单元。
-
使用Skip Connections:网络在多个编码器(encoder)深度级别使用跳跃连接(skip connections)。跳跃连接允许从编码器的深层直接传递信息到解码器(decoder),这有助于在解码器中保留更多的细节信息。
-
专用上采样解码器网络:CellViT 利用一个专门的上采样解码器网络来逐步恢复图像的空间分辨率,从而获得精确的细胞核实例分割。
-
精确的细胞核实例分割:通过上述结构,CellViT 能够生成精确的细胞核实例分割图,这意味着模型能够区分图像中每个单独的细胞核,并将它们从背景中分割出来。
-
细胞核嵌入提取:从Transformer编码器中提取细胞核嵌入(Nuclei embeddings)。这些嵌入捕获了细胞核的特征,可以用于进一步的分析,如细胞类型的分类或其他下游任务。
-
U-Net形结构:CellViT 的网络结构类似于U-Net,这是一种常见的医学图像分割模型,它具有编码器-解码器结构和跳跃连接,特别适用于需要精确定位和分割的图像分割任务。
总的来说,上图描述了一个结合了Vision Transformer和U-Net设计理念的深度学习模型,它通过特殊的网络结构来实现对细胞核的精确分割和分类。这种模型在处理医学图像,尤其是需要高精度分割的细胞核图像时,表现出了优越的性能。
四、基于DNA甲基化的分类器(SCLC-DMC),高度准确地区分小细胞肺癌(SCLC)亚型

文献概述
这篇文章讨论了一种新的方法,利用肿瘤和循环游离DNA(cfDNA)甲基化来识别具有临床意义的小细胞肺癌(SCLC)亚型。
研究团队开发了一种基于DNA甲基化的分类器(SCLC-DMC),可以高度准确地区分SCLC亚型,并且这种方法也可以应用于血液样本中的cfDNA,从而为患者提供了一种较少侵入性的亚型分配替代方案。
知识点补充:DNA甲基化
DNA甲基化是一种表观遗传修饰,涉及在DNA分子的某些碱基上添加甲基化团(-CH3)。在哺乳动物中,这种修饰通常发生在胞嘧啶的5’位置,尤其是在胞嘧啶后面紧跟着鸟嘌呤的CpG岛上。CpG岛是DNA序列中CpG位点(胞嘧啶和鸟嘌呤的组合)富集的区域,这些区域在基因组中通常与基因的启动子和调控区域相关联。
DNA甲基化的作用包括:
-
基因调控:甲基化可以调控基因的表达。在许多情况下,基因启动子区域的甲基化与基因沉默相关联,即转录活性的降低或关闭。
-
胚胎发育:在胚胎发育过程中,DNA甲基化模式的改变对于细胞分化和组织发育至关重要。
-
X染色体失活:在女性中,一条X染色体会发生甲基化,导致其失活,以保证剂量补偿效应,即保证两个性别的个体在X染色体基因表达上的平衡。
-
遗传印记:某些基因的表达依赖于它们是从父亲还是母亲那里继承的,这种单亲遗传表达模式与DNA甲基化有关。
-
肿瘤发生:异常的DNA甲基化模式与肿瘤的发生发展有关。肿瘤抑制基因的过度甲基化可能导致它们的沉默,从而促进肿瘤的发展。
-
细胞命运和多能性:在干细胞中,DNA甲基化模式对于维持其多能性(即分化成多种细胞类型的能力)至关重要。
DNA甲基化的建立和去除是由一组特定的酶来控制的,包括DNA甲基转移酶(DNMTs)。这些酶可以将甲基团从活性形式的S-腺苷甲硫氨酸(SAM)转移到DNA上的胞嘧啶碱基上。
在研究和临床应用中,DNA甲基化可以作为生物标志物,用于疾病诊断、预后评估和治疗反应监测。例如,在上文提到的小细胞肺癌(SCLC)研究中,研究人员利用DNA甲基化模式来区分不同的肿瘤亚型,这有助于个性化治疗策略的制定。
文章的主要发现和方法包括:
-
小细胞肺癌亚型识别:通过RNA测序(RNA-seq)和预测模型,可以评估小细胞肺癌的亚型。
-
DNA甲基化特征:不同亚型的小细胞肺癌具有特定的DNA甲基化模式。
-
组织和液体活检:研究展示了可以从组织和液体活检中通过DNA甲基化来区分SCLC亚型。
-
液体活检的优势:提供了一种较少侵入性的患者亚型分配替代方案。
-
研究方法:使用基因组范围的简化表示双硫仑测序(RRBS)技术,结合机器学习方法,开发了一种高度准确的DNA甲基化基础分类器。
-
临床样本分析:研究分析了两个独立队列,共包括179名SCLC患者。
-
RNA-seq和RRBS数据:生成了RNA-seq和RRBS数据,并使用机器学习方法来训练和验证分类器。
-
cfDNA分类器:开发了一个针对cfDNA的分类器(cfDMC),并展示了SCLC表型在疾病进展期间的演变。
-
治疗指导:这些数据建立了肿瘤和cfDNA甲基化可以用于识别SCLC亚型,并可能指导精准SCLC治疗的观点。
-
研究意义:这项研究提供了一种新的生物标志物,有助于小细胞肺癌的个性化治疗,并且可以用于临床治疗中的纵向跟踪。
文章强调了DNA甲基化在小细胞肺癌亚型识别中的潜力,并展示了如何利用这一生物标志物来指导治疗决策。此外,研究还探讨了不同SCLC亚型的全球甲基化水平差异,以及与甲基化相关的基因表达差异。研究结果为小细胞肺癌的精准医疗提供了重要的生物标志物,并为未来的临床试验和治疗策略的开发奠定了基础。
重点关注
Figure 1展示了小细胞肺癌(SCLC)的检测和分类方法,包括以下几个部分:
A. 基于DNA甲基化的SCLC检测的ROC分析:
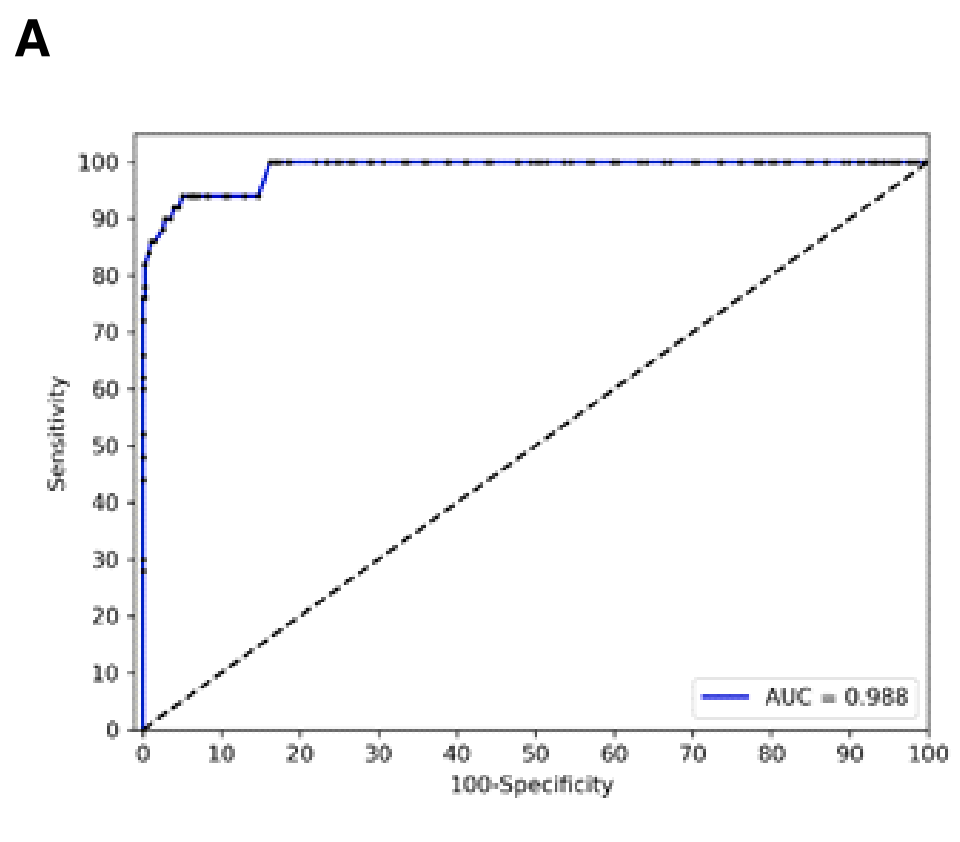
这部分展示了使用基于DNA甲基化的测试来从血浆中检测SCLC的接收者操作特征(ROC)曲线分析。ROC曲线是一种用来评估分类器性能的工具,曲线下面积(AUC)越接近1,表示测试的准确性越高。在这项研究中,检测的AUC为0.988,表明该测试具有极高的准确性。
B. 基于RNA-seq的SCLC分类的预测模型:
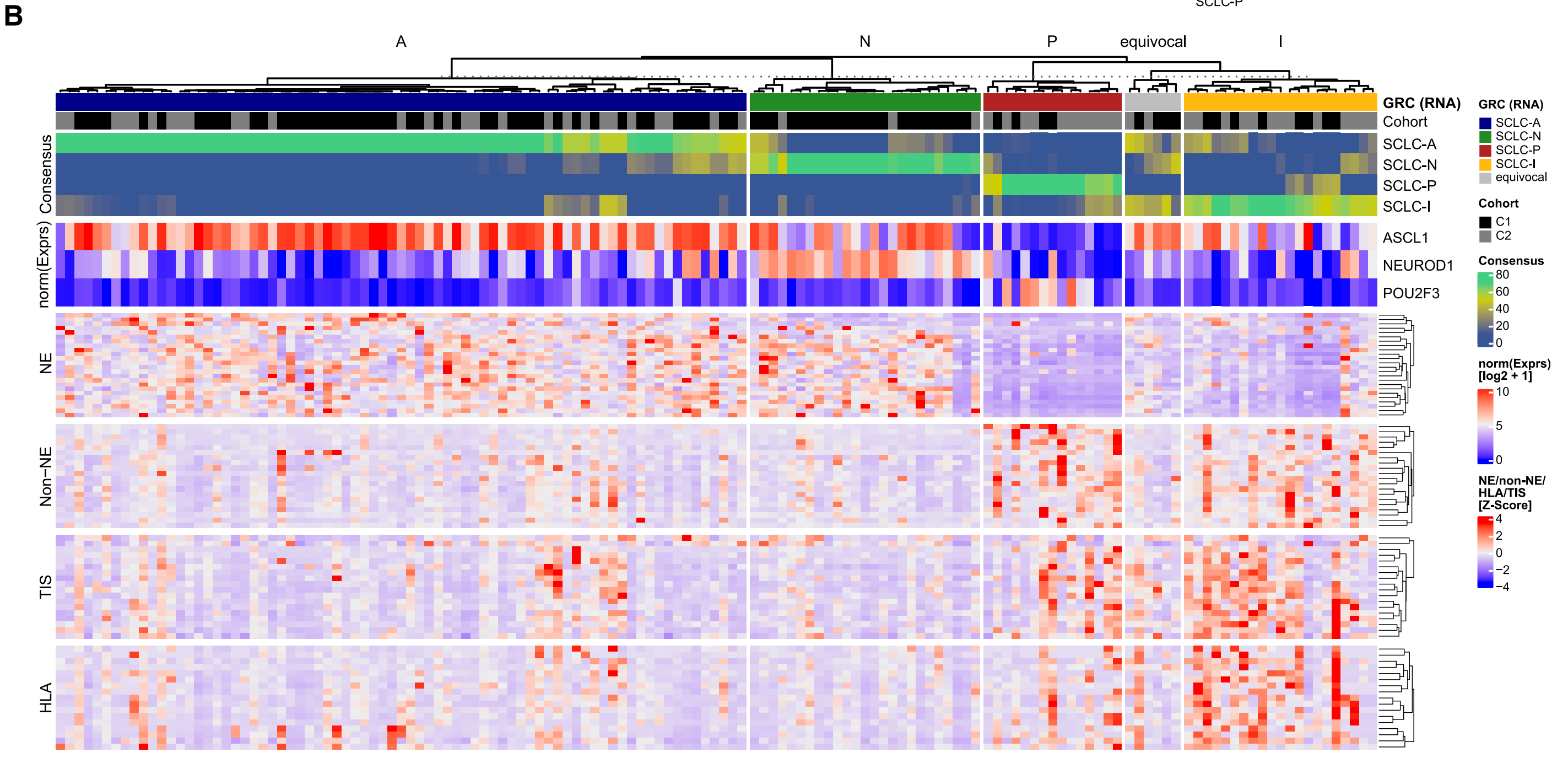
研究者开发了一个基于RNA测序数据的基因比率分类器(Gene Ratio Classifier; GRC),用于对SCLC进行亚型分类。当超过50%的模型达成共识时(即共识阈值R50),可以确定一个亚型;如果共识不足50%,则样本被归类为“不确定”。
此外,该部分还展示了三个转录因子ASCL1(SCLC-A亚型)、NEUROD1(SCLC-N亚型)和POU2F3(SCLC-P亚型)的表达情况,这些因子在两个独立队列中进行了标准化处理。还展示了与神经内分泌和非神经内分泌(Non-NE)以及肿瘤炎症(TIS)相关的基因,以及HLA基因的表达情况。
C. 免疫细胞浸润估计:
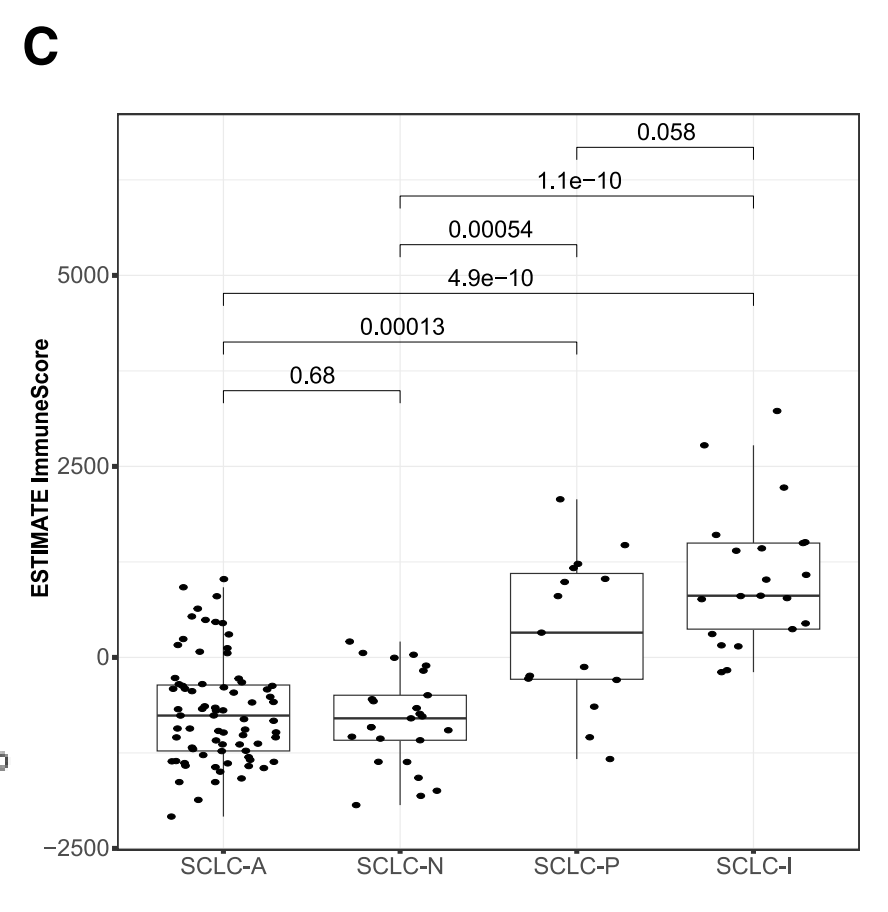
使用RNA-seq数据和ESTIMATE算法估计了免疫细胞在肿瘤中的浸润情况。箱线图显示了中位数(粗线)、第一四分位数和第三四分位数(箱子),以及四分位间距的1.5倍(须)。这有助于了解不同SCLC亚型的免疫微环境。
D. SCLC亚型共识异质性的特征:
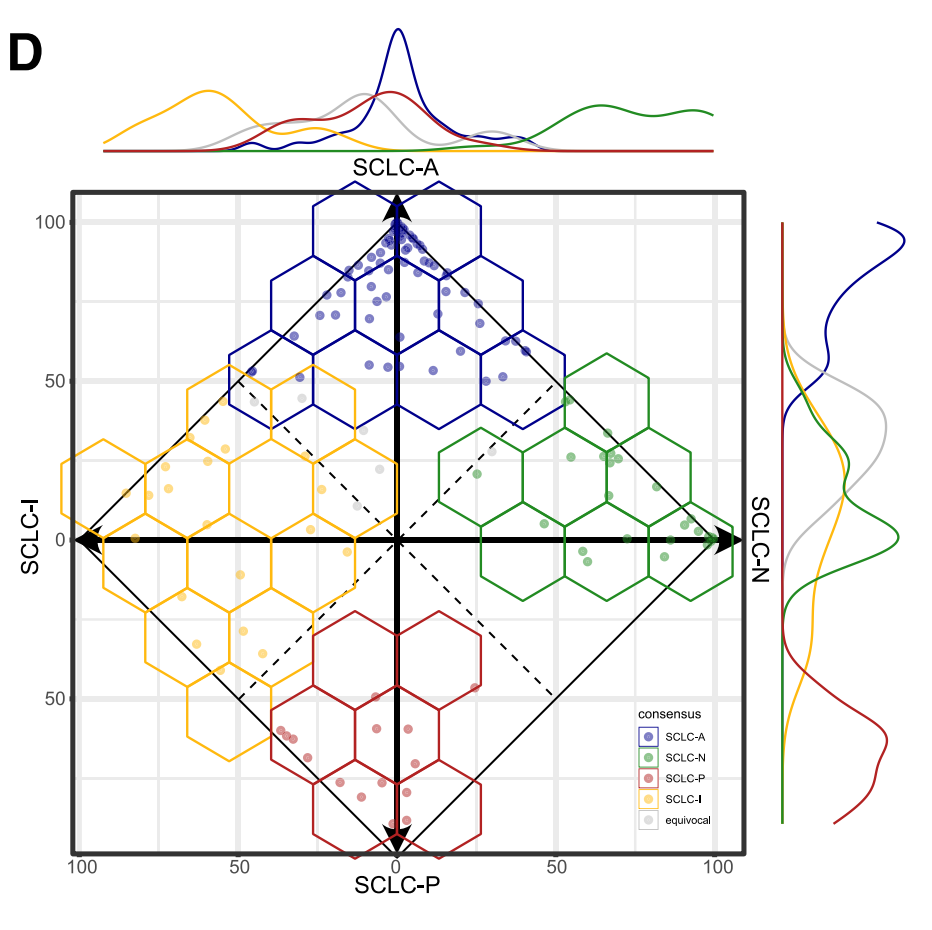
这部分展示了不同SCLC亚型之间的共识一致性值,以及它们在不同亚型中的分布。通过绘制每个亚型的共识分数,可以展示亚型之间的重叠情况。线条图显示了亚型在整个轴上的分布情况。使用Wilcoxon检验计算了不同组之间的p值,以评估统计学差异。
整体来看,Figure 1提供了SCLC检测和分类的综合视图,包括基于DNA甲基化和RNA表达的分类方法,以及免疫细胞浸润和亚型间异质性的信息。这些结果有助于更好地理解SCLC的生物学特性,并为临床治疗提供潜在的生物标志物。
五、医学图像分析中,机器学习算法性能度量标准的选取方式

文献概述
这篇文章是发表在《Nature Methods》2024年2月刊上的一篇关于医学图像分析中度量标准验证的视角文章。
文章标题为"Metrics Reloaded: recommendations for image analysis validation",作者们指出在机器学习(ML)算法验证中存在的问题,尤其是在生物医学图像分析领域,常用的性能度量标准往往不能反映领域内的兴趣点,因此无法充分衡量科学进步,并阻碍了ML技术向实践的转化。
为了解决这一问题,文章提出了一个名为"Metrics Reloaded"的全面框架,旨在指导研究人员在问题意识的驱动下选择合适的度量标准。该框架由一个大型国际联盟通过多阶段Delphi过程开发,基于一种新颖的概念——问题指纹(problem fingerprint),这是一种结构化的表示给定问题的方式,捕获了与度量标准选择相关的所有方面,从领域兴趣到目标结构的属性、数据集和算法输出。
"Metrics Reloaded"框架的目标是图像分析问题,这些问题可以被解释为图像、对象或像素级别的分类任务,即图像级别分类、对象检测、语义分割和实例分割任务。为了改善用户体验,作者们还开发了一个在线工具来实现该框架。

文章强调,尽管ML方法论在应用领域中趋于融合,但"Metrics Reloaded"促进了验证方法论的融合。文章展示了该框架在多种生物医学用例中的适用性。
作者们还讨论了度量标准的三个主要陷阱:问题类别选择不当、度量标准选择不当和度量标准应用不当。文章通过问题指纹概念,指导用户通过度量选择和应用的过程,同时提高对相关陷阱的认识。此外,文章还提出了一系列关键的设计决策,以确保度量标准的适当选择。
最后,文章呼吁更广泛的传播"Metrics Reloaded"框架,以提高生物医学成像研究的质量,并提出了定期更新框架以反映该领域最新发展的计划。作者们希望该框架和开发过程能激发科学界就成像研究中的推荐标准和良好实践进行更广泛的讨论,并最终提高研究质量。
重点关注
Fig. 1展示了"Metrics Reloaded"框架的贡献,该框架旨在解决与度量标准相关的常见问题,并提供了一种系统化的方法来选择和应用适当的度量标准。
a部分:问题动机
这里指出了与度量标准相关的三个常见问题:
- 不恰当的问题类别选择:例如,将对象检测(ObD)与语义分割(SemS)混淆(左上角)。这可能导致选用不适合的度量标准。
- 度量标准选择不当:例如,忽略了结构的小尺寸,这可能导致选用的度量标准无法准确反映算法性能(右上角)。
- 度量标准应用不当:例如,不适当的聚合方案,可能在数据聚合时未考虑数据的层次结构,这可能导致度量结果的偏差(底部)。
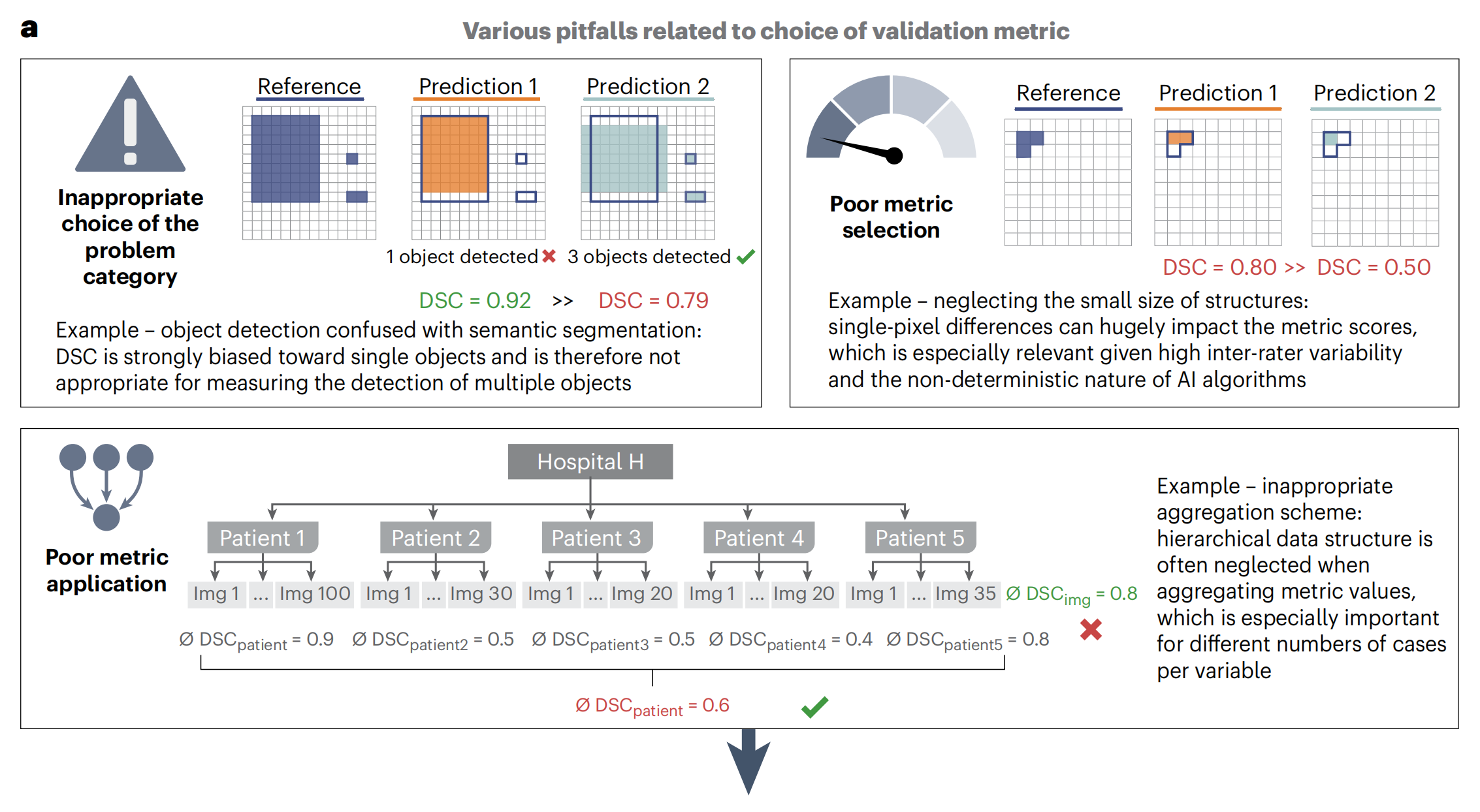
这些问题用方框标出,"∅"表示平均DSC(Dice相似系数)值。绿色度量值表示良好的度量标准,而红色则表示较差的度量标准。绿色勾号表示度量标准的期望行为,红色叉号表示不希望的行为。
b部分:Metrics Reloaded框架如何解决这些问题
- 问题指纹概念:框架基于一个新的概念——问题指纹。这是对给定的生物医学问题的有结构的表示,捕获了与度量标准选择相关的所有属性。根据问题指纹,"Metrics Reloaded"指导用户选择和应用度量标准,并提醒用户注意相关的陷阱。
- 框架实例化:展示了该框架在常见生物医学用例中的应用,证明了其广泛的适用性。
- 在线工具:提供了一个公开可用的在线工具,以促进框架的应用。

此外,文中提到了对参考文献27的改编,遵循了创作共用许可CC BY 4.0。第二个输入图像是根据创作共用许可CC BY 4.0从dermoscopedia(参考文献58)复制的;第四个输入图像是经过美国物理学家医学会(参考文献59)许可复制的。
总体而言,Fig. 1强调了在度量标准选择和应用过程中需要考虑的关键问题,并介绍了"Metrics Reloaded"框架如何通过问题指纹和在线工具来解决这些问题,从而提高度量标准的准确性和适用性。
六、AI+蛋白组学,实现早期非小细胞肺癌(NSCLC)的诊断以及肺部小结节的良恶性区分

文献概述
这篇文章的标题是《Serum Protein Fishing for Machine Learning-Boosted Diagnostic Classification of Small Nodules of Lung》,发表在《ACS Nano》2024年第18卷,第4038-4055页。
文章的核心内容是关于如何提高对肺部小结节良恶性诊断的准确性。
作者们开发了一种基于血清蛋白质捕捞的纳米光谱库方法(ProteoFish),结合机器学习技术,用于早期非小细胞肺癌(NSCLC)的诊断以及区分肺部小结节的良性和恶性。
这项技术通过使用来自297名临床受试者的血清样本,建立了一个广泛的NSCLC蛋白质库。在测试了5种特征提取算法和六种机器学习模型后,选择了Lasso算法来选择15个关键蛋白质面板,并使用随机森林模型进行诊断分类。
研究结果表明,随机森林分类器在区分良性和恶性小结节的诊断中达到了91.38%的准确率,这一表现优于现有的临床检测方法。通过将机器学习与蛋白质组学数据分析相结合,识别出的关键蛋白质面板可以为从血清中进行多重蛋白质生物标记物捕捞提供见解,有助于癌症筛查,并解决当前临床诊断分类小结节的挑战。
文章还讨论了肺癌的全球死亡率,指出肺癌是全球癌症相关死亡的最常见原因,非小细胞肺癌(NSCLC)占肺癌死亡的85%。肺部小结节是肺部内部细胞的小型生长,它们在早期阶段彼此非常相似,因此早期检测恶性肺结节对预后至关重要。尽管近年来在肺癌诊断方面取得了显著进展,但现有的诊断工具如低剂量计算机断层扫描(LDCT)等,虽然能够检测到大多数早期结节,但无法区分良性和恶性肿瘤,导致高假阳性率和过度医疗治疗。
文章进一步介绍了血清蛋白质捕捞技术(ProteoFish),这是一种利用血液中的蛋白质动态特征来反映身体变化的技术。与抽吸活检相比,从患者身上采集血液是微创的,并且患者依从性高。然而,血清或血浆的复杂性和蛋白质浓度的广泛动态范围使得使用质谱(MS)或其他手段在高丰度蛋白质存在下检测低丰度蛋白质非常困难。
最后,文章提出了未来的研究方向,包括使用算法整合来自多个实验室的蛋白质组学数据,以及开发能够诊断多种癌症类型的算法。作者们认为,通过结合多组学数据(如基因组学、代谢组学、蛋白质组学等)和医学影像以及人工智能,可以实现对肺部恶性和良性结节更准确的分类。
这篇文章为肺癌的早期诊断和治疗提供了新的思路和方法,有望改善肺癌患者的预后,并减少不必要的过度治疗。
重点关注
Figure 1 展示了使用 ProteoFish 技术建立纳米光谱库和发现非小细胞肺癌(NSCLC)生物标志物的详细工作流程。
该流程分为四个阶段:
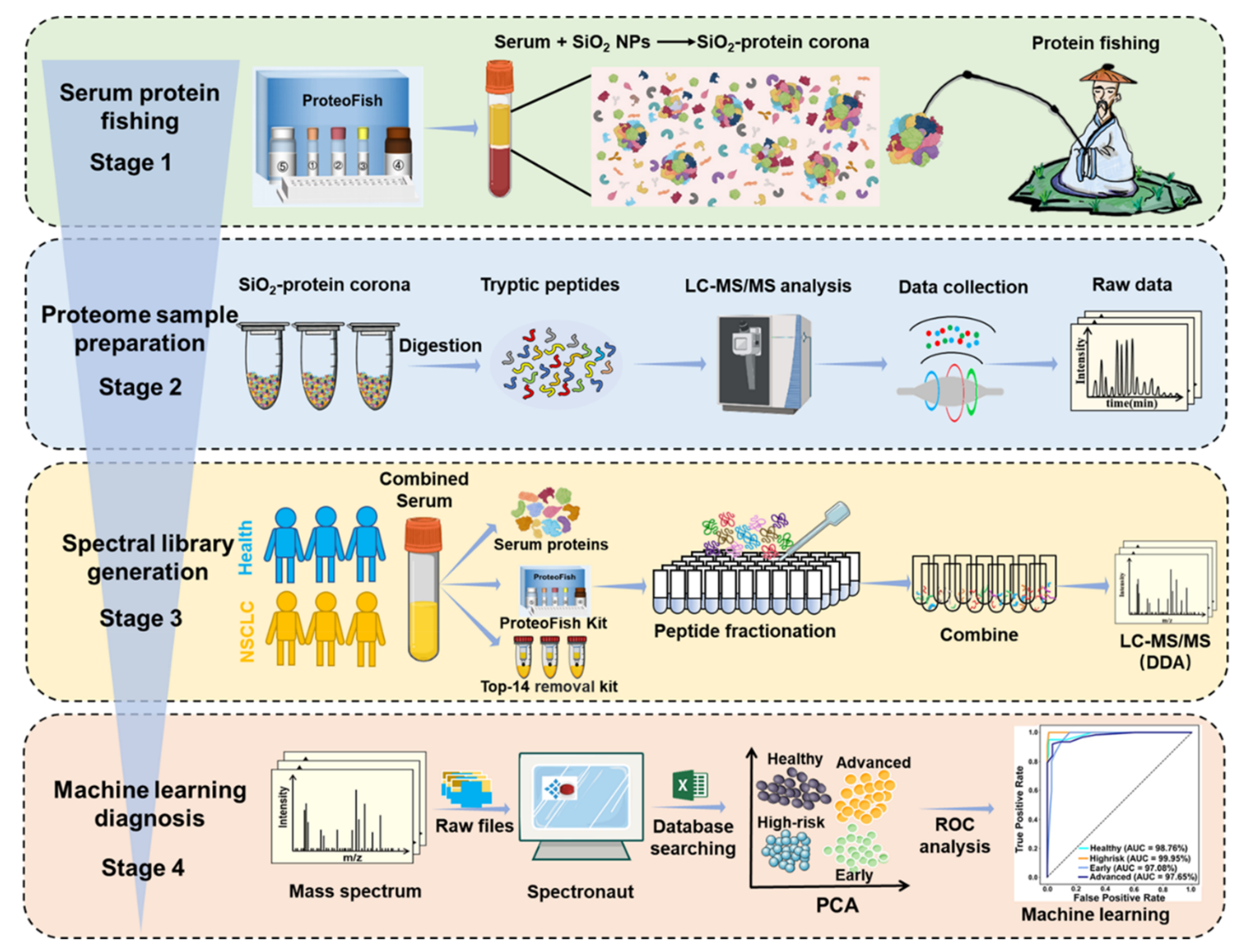
第一阶段(Stage 1):血清蛋白质捕捞
- 利用 ProteoFish 技术通过形成二氧化硅(SiO2)-蛋白质冠(protein corona)进行血清蛋白质捕捞。
- SiO2 纳米粒子能够从血清中富集低丰度蛋白质,为下一步的粒子上消化(on-particle digestion)做准备。
第二阶段(Stage 2):SiO2-蛋白质冠的预处理
- 通过粒子上消化,对 SiO2-蛋白质冠进行预处理。
- 使用 Orbitrap Exploris 480 进行液相色谱-串联质谱(LC-MS/MS)数据收集,以获取蛋白质的详细信息。
第三阶段(Stage 3):全面光谱库的生成
- 通过高pH反相色谱分离,为数据非依赖性采集(DIA)基础的蛋白质组分析生成全面的光谱库。
- 光谱库包括血清光谱库、基于ProteoFish试剂盒的库和基于去除前14种蛋白质试剂盒的光谱库。
第四阶段(Stage 4):DIA基础的蛋白质组分析
- 使用 Spectronaut 软件和基于机器学习的模型进行 DIA 基础的蛋白质组分析。
- 通过关键蛋白质的选择进行 NSCLC 的诊断分类。
整个工作流程的核心在于利用 SiO2 纳米粒子捕捞血清中的低丰度蛋白质,这些蛋白质可能与 NSCLC 的发展有关。通过富集这些低丰度蛋白质并使用先进的质谱技术进行分析,研究人员可以更深入地了解肺癌的蛋白质组学特征,并识别出可能的生物标志物。然后,通过机器学习模型对这些蛋白质进行分析,以提高 NSCLC 的诊断准确性。这种方法为早期发现和治疗 NSCLC 提供了一种潜在的、有效的工具。


















 1819
1819

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









