






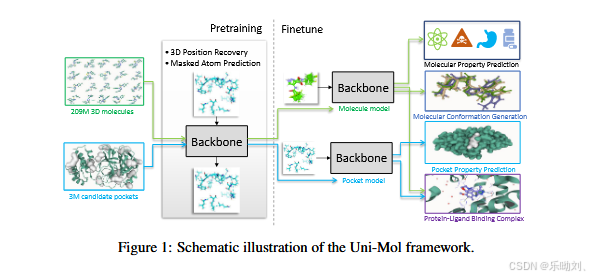
- 背景与动机
- 表示学习在许多领域广泛应用,但药物设计领域的分子表示学习面临挑战,现有方法多将分子视为一维或二维结构,难以有效利用三维信息,而分子和药物的性质多由三维结构决定。
- Uni - Mol 框架
- 骨干网络:基于 Transformer,能处理三维数据,引入多种改进以编码三维位置,包括基于原子对欧几里得距离和高斯核的位置编码,维护原子和成对表示并通过特定方式更新以增强三维空间表示,还能通过 SE (3) - 等变头预测三维位置。
- 预训练:使用包含 2.09 亿分子构象和 300 万候选蛋白口袋的大规模数据集,通过三维位置恢复和掩蔽原子预测任务,采用随机位置和噪声范围等技术,让模型学习三维结构信息。
- 微调:根据任务类型和是否涉及蛋白质或配,分为非三维预测任务(如分子性质预测等,可利用 [CLS] 表示或原子平均表示微调)、分子或口袋的三体维预测任务(如分子构象生成,在 Uni - Mol 中变为构象优化任务)、蛋白质 - 配体对的三维预测任务(采用基于评分函数的优化方法预测复合物结构)。
选择 Transformer 作为骨干网络
在分子表示学习(MRL)中,常见的骨干网络模型有图神经网络(GNN)和 Transformer。由于 GNN 通常使用局部连接图来表示分子,缺乏捕捉原子间长程相互作用的能力,而长程相互作用在 MRL 中很重要,所以 Uni - Mol 选择 Transformer 作为骨干网络,因为它能全连接节点 / 原子,从而学习可能的长程相互作用。
对标准 Transformer 进行修改以处理 3D 数据
虽然有一些工作将 Transformer 扩展到 3D 数据领域,但由于加入了复杂组件(如张量场网络),导致速度比标准 Transformer 慢。考虑到在大规模数据集上进行预训练的成本,需要一个高效的骨干网络。因此,基于带有前置层归一化(Pre - LayerNorm)的标准 Transformer,进行了一些高效且必要的修改,使其能够将 3D 位置作为输入和输出。
Uni - Mol 骨干网络架构概述
- 输入和表示
- 有两个输入:原子类型和原子坐标。
- 模型中维护了两种表示:原子表示和成对表示。
- 原子表示通过嵌入层(Embedding layer)由原子类型初始化;成对表示由根据原子坐标计算出的不变空间位置编码初始化,该编码基于原子间的成对欧几里得距离,对全局旋转和平移是不变的,两种表示在自注意力模块中相互交互。
- 对 3D 位置进行编码
- Transformer 具有置换不变性,没有位置编码无法区分输入位置。3D 空间位置是连续值,与 NLP/CV 中的离散位置不同,且位置编码过程需在全局旋转和平移下保持不变。
- 对现有的 3D 空间位置编码进行了基准测试后,选用了一种简单有效的方法:原子对的欧几里得距离加上考虑成对类型的高斯核。
- 在 Transformer 中维护了一个成对层面的表示,通过自注意力机制中多头查询 - 键(Query - Key)乘积的结果进行原子到成对的信息传递来更新成对表示,还通过将成对表示作为自注意力中的偏差项来实现成对到原子的信息传递,这些操作简单且额外成本可忽略不计,基准测试证明了其效率和有效性。

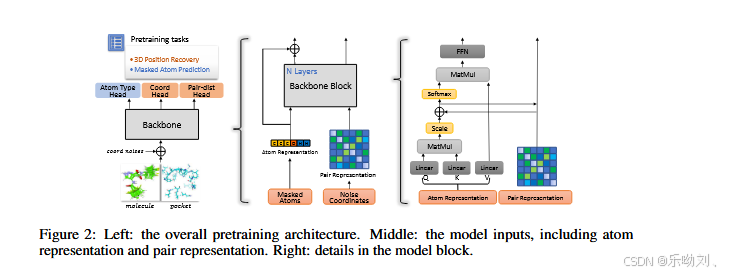
左部分:整体预训练架构
- 输入(Inputs)
- 有两个输入源:分子(molecule)和口袋(pocket)。
- 输入包括原子类型(Atom Type)、坐标(Coord)和成对距离(Pair - dist)。
- 骨干网络(Backbone)
- 输入经过骨干网络处理。
- 预训练任务(Pretraining tasks)
- 包括两个预训练任务:3D 位置恢复(3D Position Recovery)和掩蔽原子预测(Masked Atom Prediction)。
中部分:模型输入,包括原子表示和成对表示
- 原子表示(Atom Representation)
- 由原子类型初始化。
- 成对表示(Pair Representation)
- 由不变空间位置编码初始化,该编码基于原子坐标计算得出。
右部分:模型块细节
- 骨干网络块(Backbone Block)
- 由 N 层组成。
- 每层包括线性层(Linear)、矩阵乘法(MatMul)、缩放(Scale)和 Softmax 操作。
- 处理原子表示和成对表示。
- 前馈网络(FFN)
- 用于进一步处理原子表示和成对表示。
3D 位置预测
- 有了 3D 空间位置编码和成对表示,模型能够学习到良好的 3D 表示,但还缺乏直接输出坐标的能力。为此,引入了一个 SE (3) - 等变头来预测增量位置。该头类似于 EGNN 中的位置更新过程,但由于 Uni - Mol 只在最后一层更新 3D 位置,所以效率更高。作者通过基准测试证明了 Uni - Mol 中的方法更好。
- Uni - Mol 的骨干网络可以被任何能够将 3D 位置作为输入和输出的 SE (3) 模型所替代,但考虑到预训练成本,作者仅对标准 Transformer 模型进行了一些简单且必要的修改。
预训练
- 大规模数据集构建
- 分子预训练数据集:基于多个公开数据集,经归一化和去重后含约 1900 万个分子,使用 ETKGD 结合默克分子力场在 RDKit 中优化,为每个分子随机生成 11 种构象,共 2.09 亿种构象。
- 蛋白口袋预训练数据集:源自蛋白质数据银行(RCSB PDB)的 18 万个蛋白质 3D 结构,经数据清理(添加缺失部分)、用 Fpocket 检测结合口袋、按残基数量筛选后,得到 320 万个候选口袋数据集。
- 预训练策略
- 自监督任务设计:为让模型学习 3D 结构信息,设计 3D 位置恢复自监督任务,因 3D 位置连续不能用类似 BERT 的掩蔽方式,而是用随机位置作损坏输入,训练模型预测正确位置。
- 处理技术
- 重新分配:给定 m 个原子和 m 个随机位置,用高效贪心算法找次优重新分配,使随机与真实位置差值减小,但最终发现无需重新分配。
- 噪声范围:限制随机位置空间,经测试发现 r = 1 Å 效果好。
- 基于损坏输入的恢复操作
- 用两个额外头恢复正确位置,包括基于成对表示预测损坏原子对的欧几里得距离(成对距离预测)和基于 SE (3) - 等变坐标头预测损坏原子坐标(坐标预测)。
- 对损坏原子类型掩蔽并预测正确类型。用特殊原子 [CLS](坐标为所有原子中心)代表分子 / 口袋,两预训练模型均用此自监督任务,预训练配置见附录 C,策略选择基于广泛基准测试
微调阶段
- 总体原则
- 与预训练保持一致的数据预处理,分子可利用随机构象增强,有原子坐标任务则直接用,跳过 3D 构象生成。
- 按任务类型分类微调
- 非 3D 预测任务:如分子性质预测等,用 [CLS] 表示或原子平均表示加线性头微调,口袋 - 分子对任务则连接 [CLS] 表示后微调。
- 分子或口袋的 3D 预测任务:如分子构象生成,在 Uni - Mol 中变为构象优化任务,模型学习从生成构象到标记构象的映射,输出构象由 SE (3) - 等变头端到端生成。
- 蛋白质 - 配体对的 3D 预测任务:预测复合物结构,因直接用 SE (3) - 等变头不稳定,采用基于评分函数的优化方法,先获分子和口袋表示并连接输入解码器微调学习原子对距离,以距离矩阵为评分函数随机放置配体后反向传播优化坐标,比传统对接工具快约 100 倍,更多细节在附录
实验
分子性质预测
- 数据集与设置:
- 数据集选用:采用分子性质预测常用基准 MoleculeNet,涵盖从量子力学到生理学等不同分子性质相关数据集。遵循 GEM 的做法,使用支架分割方法,并依据 3 个随机种子的结果报告均值和标准差。
- 对比基线:
- 将 Uni - Mol 与多种基线对比,包括监督学习和预训练方法两类基线。
- 监督学习的图神经网络方法有 D - MPNN 和 AttentiveFP;预训练方法包含 N - gram、PretrainGNN、GROVER、GraphMVP、MolCLR、GEM,其中 N - gram 是将图中节点嵌入后按短路径组合成图表示,下游任务预测器采用随机森林和 XGBoost。
- 实验结果:
- 表 1 和表 2 展示了 Uni - Mol 与各竞争基线的实验结果(最佳结果加粗显示),多数基线结果源于 GEM 论文,GraphMVP 和 MolCLR 结果分别取自其各自论文,因 MolCLR 数据分割设置不同(未考虑手性),按其他基线相同设置重新运行了它。
- 总结来看:
- 总体上,Uni - Mol 在几乎所有下游数据集上优于基线方法。
- 在溶解度(Lipo)、自由能(FreeSolv)及量子力学(QM7、QM8、QM9)性质预测任务中,Uni - Mol 显著优于基线,鉴于 3D 信息在这些性质中至关重要,表明 Uni - Mol 能学到比其他基线更好的 3D 表示。
- Uni - Mol 在 SIDER 数据集上未超越当前最优水平,经调查发现,它无法为 SIDER 数据集中许多分子(如天然产物和肽)生成 3D 构象,因缺少 3D 信息而无法优于其他方法。
- 总之,通过在预训练中更好利用 3D 信息,Uni - Mol 在几乎所有性质预测任务中表现优于此前所有分子表示学习(MRL)模型。
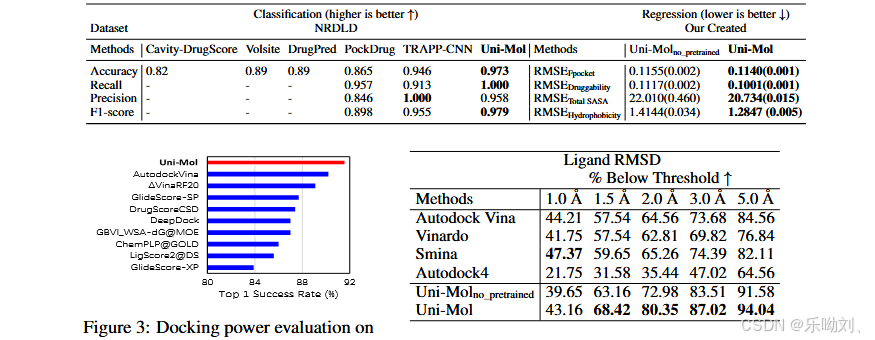
蛋白质 - 配体结合姿态预测
- 数据集与设置:
- 任务重要性及 Uni - Mol 的处理方式:蛋白质 - 配体结合姿态预测是药物设计中极为重要的任务,Uni - Mol 结合分子和口袋预训练模型学习基于距离矩阵的评分函数,进而优化复合物构象。
- 数据集选用:参照以往研究,将 CASF - 2016 作为测试集,微调时的训练数据采用 PDBbind General set v.2020(含 19443 个复合物),为检验泛化能力,进一步过滤掉与测试集(CASF - 2016)中相似的训练复合物,具体是过滤掉蛋白质序列相似度(MMSeqs2 相似度高于 40%)和分子相似度(指纹相似度高于 80%)都高的复合物,过滤后剩下 18404 个复合物。
- 两个基准测试:
- 对接能力(Docking power):这是 CASF - 2016 中衡量评分函数能力的默认指标,测试评分函数能否从一组诱饵构象中区分出真实结合姿态,CASF - 2016 为每个真实结合姿态提供 50 - 100 个同一配体的诱饵构象,用评分函数对它们进行排序,期望真实结合姿态排在首位。
- 结合姿态准确性(Binding pose accuracy):采用半柔性对接设置,即保持口袋构象固定,配体构象完全灵活,通过评估预测结果与真实结果之间的均方根偏差(RMSD)来衡量,按照以往做法,以低于预定义 RMSD 阈值的结果百分比作为评价指标。
结论
- 本文为扩大分子表示学习(MRL)的应用范围和表示能力,提出了 Uni - Mol 这一首个通用大规模 3D MRL 框架,它由三部分组成:处理 3D 数据的基于 Transformer 的骨干网络、分别学习分子和口袋表示的两个大规模预训练模型、针对各类下游任务的微调策略。实验表明 Uni - Mol 在多个下游任务尤其是 3D 空间任务中优于现有 SOTA。
- 提出三个潜在的未来发展方向:
- 优化微调时预训练模型的交互机制:当前 Uni - Mol 版本中预训练口袋模型和预训练分子模型之间的交互较简单,有很大改进空间。
- 训练更大规模的 Uni - Mol 模型:更大的预训练模型往往性能更好,值得在更大数据集上训练大模型。
- 打造更多高质量基准测试:药物设计领域虽应用众多,但缺乏高质量公共数据集,很多现有数据集因数据质量低无法满足实际需求,高质量基准测试将显著推动药物设计领域发展。

















 853
853

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









