










Uni-Mol Docking V2 模型是 Uni-Mol Docking 的升级版本,作者来源于深度势能 AI for Sence 团队。Uni-Mol Docking V2 在 PoseBusters 基准测试中,77% 以上的配体预测结合姿态的 RMSD 值小于 2.0 Å,75% 以上通过了所有质量检查,显著优于先前版本的 62% 准确率, 解决了先前机器学习模型中出现的手性倒置和空间冲突等问题。V2 版本使用来自 MOAD 数据库的蛋白质-配体结合数据进行训练,并采用特定流程来准备蛋白质数据,包括添加正确的氢原子、质子化信息以及补全缺失的重原子和残基。
一、背景介绍
Uni-Mol Docking V2 是由深势科技 AI for Sence 团队基于 Uni-Mol Docking 升级的版本,来源于文章:《Uni-Mol Docking V2: Towards Realistic and Accurate Binding Pose Prediction》。文章链接:https://arxiv.org/pdf/2405.11769 。该文章在 2024 年 5 月 20 日发表于 arXiv 上。

UniMol Docking 的 V1 版本,是Uni-Mol 文章发布的时候的蛋白-小分子3D位置预测子任务,详见文章 《UNI-MOL: A UNIVERSAL 3D MOLECULAR REPRESENTATION LEARNING FRAMEWORK》。
近年来,机器学习(ML)方法作为分子对接的有力替代方案,因其有可能在不产生高昂计算成本的情况下提供高精度预测而受到关注。然而,近期研究表明,这些 ML 模型可能过度拟合定量指标,而忽略了问题中固有的物理约束。在本研究中,作者提出了 Uni-Mol Docking V2,展示了性能的显著提升,能够精确预测 PoseBusters 基准测试中超过 77% 的配体的结合位点,且 RMSD 值低于 2.0 Å,其中超过 75% 的预测通过了所有质量检查。这一结果较之前的 Uni-Mol Docking 模型(62%)有了显著提高。值得注意的是,Uni-Mol Docking 方法生成了化学上准确的预测,避免了以往机器学习模型常见的手性反转和立体冲突等问题。此外,作者还观察到,当 Uni-Mol Docking 与更多基于物理的方法(如Uni-Dock)结合时,其在高质量预测(RMSD 值小于 1.0 Å 和 1.5 Å)和物理合理性方面表现得更为出色。作者的结果代表了人工智能在科学研究中的重要进展,采用了整体方法进行配体对接,适合用于虚拟筛选和药物设计等工业应用。Uni-Mol Docking 的代码、数据和服务可公开获取,供用户使用和进一步开发,链接为:GitHub - deepmodeling/Uni-Mol: Official Repository for the Uni-Mol Series Methods
二、模型介绍
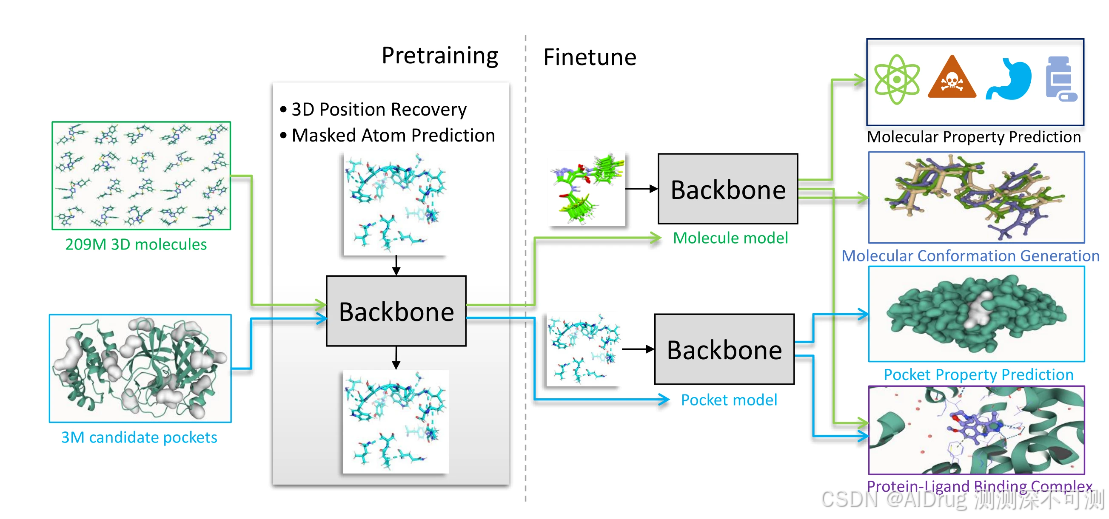
Uni-Mol Docking V2 是基于 Uni-Mol 模型开发的用于分子对接任务的模型。下图展示了 Uni-Mol 的总体框架。Uni-Mol 的框架主要包含两个模型:一个是使用 2.09 亿个分子的三维构象进行训练的分子预训练模型;另一个使用了 300 万个候选蛋白质数据进行训练的口袋预训练模型。这两个模型可以独立用于不同的任务分别进行预测,也在蛋白质-配体结合任务中结合使用。Uni-Mol 在 15 个分子性质预测任务中,有 14 个任务的预测结果优于当前最先进的技术。此外,Uni-Mol 在蛋白质-配体结合方式预测和分子构象生成等三维空间的预测结果也十分优良。Uni-Mol 建模系列描述了通用分子编码器的预训练,并展示了它们在各种二维和三维下游任务中的应用,如分子构象生成、分子属性预测和分子对接。随后,Uni-Mol 范式扩展到量子属性预测,展示了该架构学习的分子特征的质量和适用性。
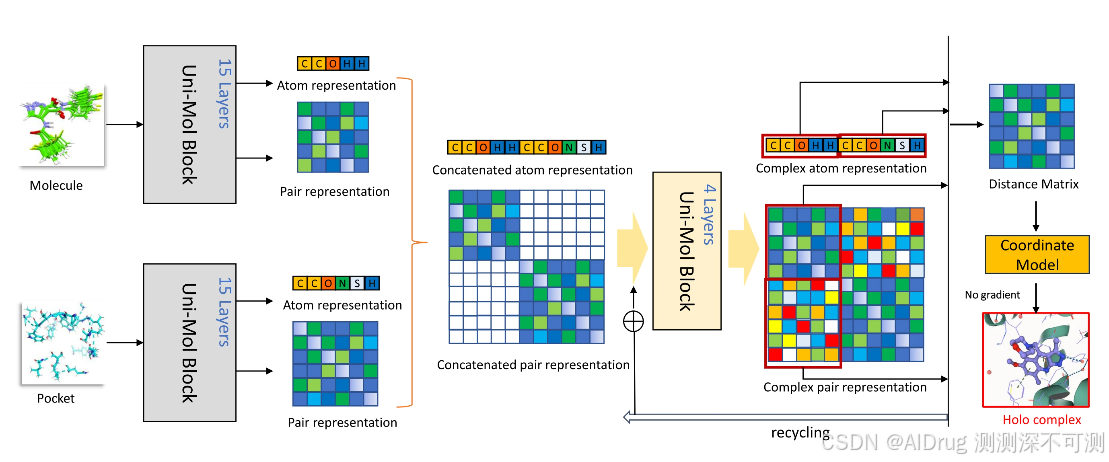
下图展示了 UniMol Docking V2 的框架,模型输入化合物和蛋白口袋到预训练的分子编码器、预训练的口袋编码器,结构使用原子表示和对表示,其中原子表示通过嵌入层由原子类型初始化,原子对表示通过原子坐标计算的不变的空间位置编码初始化。并且基于原子之间的欧几里得距离,原子对表示对全局旋转和平移具有不变性。两部分信息分别拼接到一起,最终通过联合口袋-配体模块输出预测的复合物构象。UniMol Docking V2 在 CASF-2016 基准测试中相较于传统的对接算法如 Autodock Vina 取得了优异的表现。
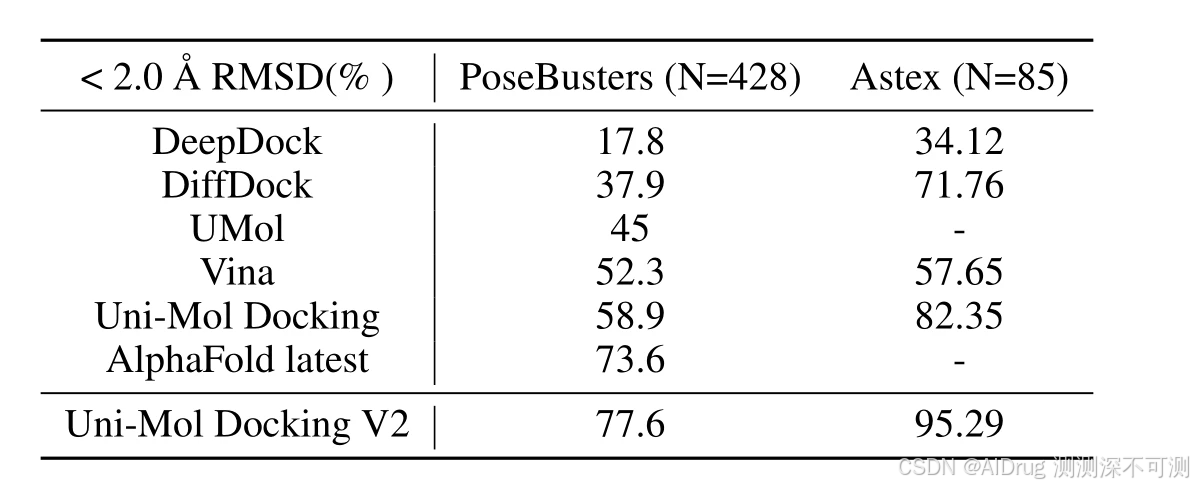
最近的研究强调了对 ML 模型预测的对接位点进行物理和化学合理性评估的必要性,并显示出尽管深度学习模型在定量指标上表现更好(如 <2.0 Å RMSD 的百分比),但它们在性能上并未显著超越传统的对接程序。在这项研究中,UniMol Docking V2 报告在 PoseBusters 数据集上取得了 <2.0 Å RMSD 指标上的最好表现。作者将这一表现归因于数据处理的优化,并在本研究中提出了标准的处理流程。下表展示了当前先进的对接方法在 PoseBusters 和 Axtes 测试集上的表现,可以看出 UniMol Docking V2 都有优异的表现。
近年来,一些研究提出了不同的解决方案以应对 ML 对接的不足。在模型方面,RFAA 提出了全原子建模,并将蛋白质折叠扩展到蛋白质、核酸、离子和配体;DiffDockpocket 和 Umol 提出通过在建模中引入蛋白质柔性来降低对晶体结构的需求;最新的 AlphaFold 报告采用了类似的方法,并在 PoseBusters 基准测试中取得了最佳结果,同时在化学准确性(立体化学)方面也有一定的改进。在结果的合理性方面,轻量级的后处理被简要探讨过,但未能完全消除不合理的结果。
因此,本研究介绍了以下三项主要成果:
1. 为之前发布的 Uni-Mol Docking 提供了一个可重复的分子对接设置,经过正确的数据集处理和可重复的结果,在 CASF-2016、PoseBusters 测试集和 Astex Diverse Set 中达到了最先进的水平,并且公布了代码。
2. UniMol Docking V2 的结果展示了在未见过数据上的性能提升,并在截至 2023 年 11 月 22 日的 PoseBusters 测试集上取得了所知的最佳结果。
3. 深度学习模型的化学准确性发生了显著变化,之前 ML 模型预测中存在的所有物理和化学问题已被修正。
作者从 MOAD 收集蛋白质-配体结合数据用于训练。蛋白质数据使用特定的处理流程进行准备,包含正确添加氢原子、质子化信息,以及补全缺失的重原子和残基。作者将数据随机划分为训练集和验证集,比例为 9:1。
Uni-Mol Docking V2 的训练从预训练的分子和口袋模型检查点开始,和 V1 版本相同。作者在 8 个 V100 GPU 上训练模型 100 个epoch,批量大小为 64,相比 V1 版本的批量大小翻倍。Uni-Mol Docking V2 的输入与之前的版本相同:已知的口袋和待对接的化学化合物。基于这些输入,口袋以配体大小的立方体格式提供,外加 10 Å 的边距(类似于现有工具如 AutoDock Vina),并且提供一个配体构象(可以是手动提供,或者使用标准的化学信息学工具从配体的 SMILES 结构自动构建)。输出是配体与目标蛋白结合后的三维姿态。通过预计算口袋特征,Uni-Mol Docking 能够高效地应用于虚拟筛选场景。
Uni-Mol Docking 与 UniDock 的结合适合用于工业应用和理性药物设计案例,其中结合口袋得到了更好的表征。UniDock 进一步允许利用来自辅因子和晶体水的信息,以进一步提高准确性。PoseBusters 测试集由 UniMol(包括先前版本和最新的 V2 版本)未见过的数据组成,因为它只包含截至 2019 年发布的选定蛋白质-配体复合物,而测试集中的结构是从 2020 年起发布的。
Uni-Mol Docking V2 的训练从预训练的分子和口袋模型检查点开始,和 V1 版本相同。作者在 8 个 V100 GPU 上训练模型 100 个epoch,批量大小为 64,相比 V1 版本的批量大小翻倍。Uni-Mol Docking V2 的输入与之前的版本相同:已知的口袋和待对接的化学化合物。基于这些输入,口袋以配体大小的立方体格式提供,外加 10 Å 的边距(类似于现有工具如 AutoDock Vina),并且提供一个配体构象(可以是手动提供,或者使用标准的化学信息学工具从配体的 SMILES 结构自动构建)。输出是配体与目标蛋白结合后的三维姿态。通过预计算口袋特征,Uni-Mol Docking 能够高效地应用于虚拟筛选场景。
Uni-Mol Docking 与 UniDock 的结合适合用于工业应用和理性药物设计案例,其中结合口袋得到了更好的表征。UniDock 进一步允许利用来自辅因子和晶体水的信息,以进一步提高准确性。PoseBusters 测试集由 UniMol(包括先前版本和最新的 V2 版本)未见过的数据组成,因为它只包含截至 2019 年发布的选定蛋白质-配体复合物,而测试集中的结构是从 2020 年起发布的。
2.1 Uni-Mol Docking V2 在 PoseBusters 基准上的结果
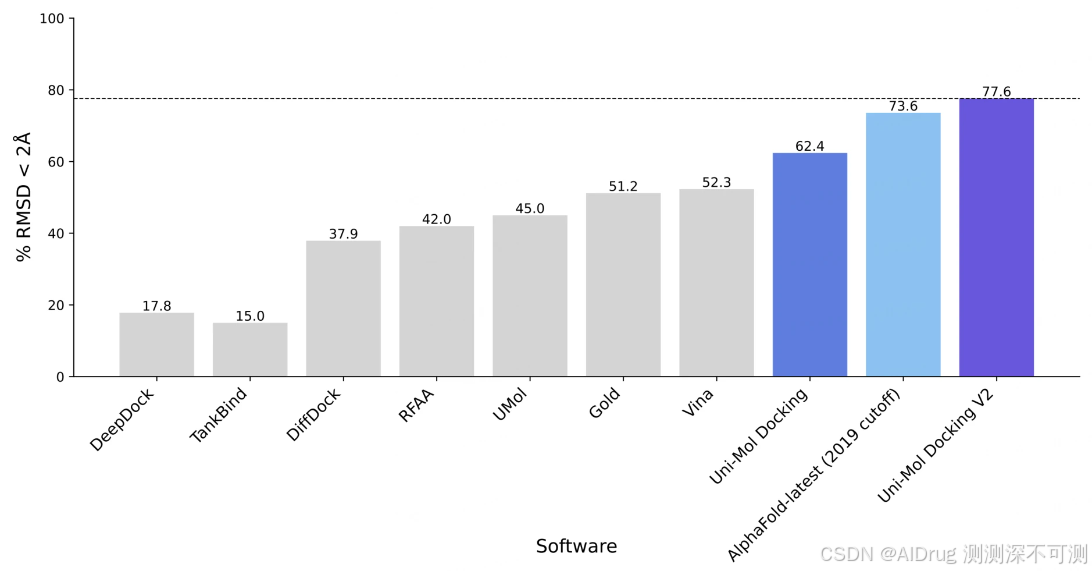
下图中,作者展示了 Uni-Mol Docking V2 版本的结果,在 PoseBusters 基准测试中,超过 77% 的配体的 RMSD 小于 2.0 Å,且超过 75% 的复合物通过了所有PoseBusters质量检查。这代表了机器学习辅助蛋白质-配体对接的新最前沿。
2.2 Uni-Mol Docking V2 在化学准确性上带来了提升
此外,作者强调 Uni-Mol Docking V2 能够生成化学上准确的预测构象,展示了没有手性反转或立体冲突的结果,这与之前的机器学习模型不同。下图展示了不同方法在化学合理性方面的表现。Uni-Mol Docking V2 的 95% 以上的预测在化学和物理上都是合理的。
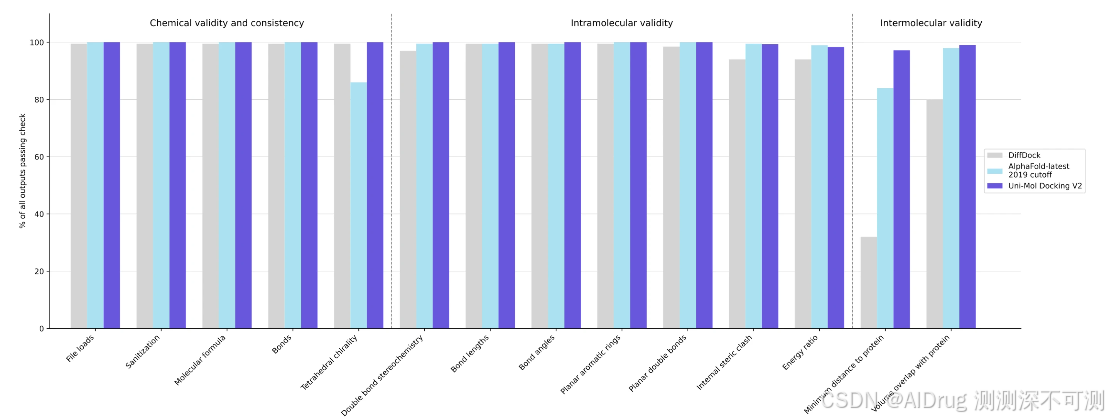
作者还报告了在高质量预测(RMSD<1.0 Å和<1.5 Å)和提高物理合理性方面的性能提升,特别是当 Uni-Mol Docking V2 与基于物理的方法(如 Uni-Dock)结合使用时。此设置提高了理性药物设计和虚拟筛选的工业应用,具有更高的整体准确性、较低的过拟合风险、更高的高质量预测比例,并且能够整合结合位点的附加信息,如辅因子和晶体水。
2.3 案例研究
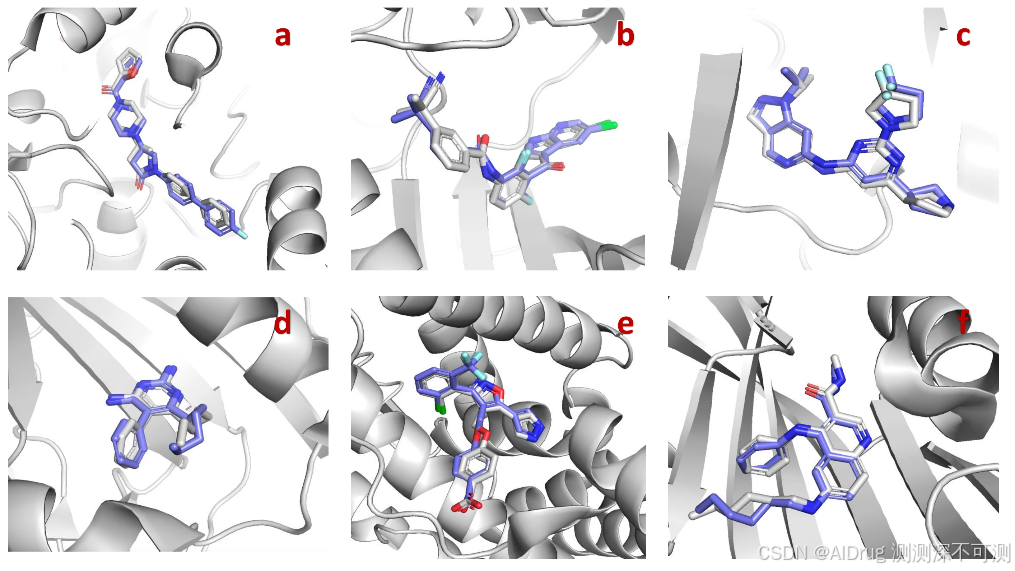
作者在多个重要的生物体系里验证了 Uni-Mol Docking V2 的预测结果。如下方所示,Uni-Mol Docking V2 展示出与实验精度接近的预测效果。图中蓝色为预测结果,灰色为实验结果。(a-f:7PRM、8C7Y、7R9N、7XI7、7NP6、7N03)
三、Uni-Mol-Docking V2 评测
3.1 下载数据集
项目提供训练数据集,保存在 senodo 上,链接为:https://zenodo.org/records/11191555,文件内容如下所示:
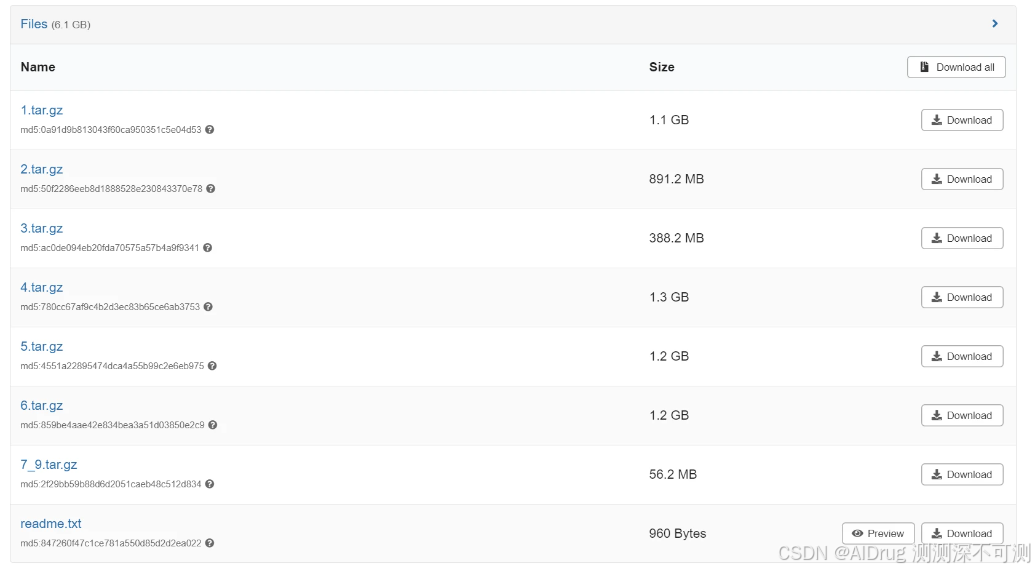
下载训练集所有的压缩文件以及说明文件(readme.txt),上传到 ./data 文件夹中,通过下面命令解压:
tar -xzvf 1.tar.gz
tar -xzvf 2.tar.gz
tar -xzvf 3.tar.gz
tar -xzvf 4.tar.gz
tar -xzvf 5.tar.gz
tar -xzvf 6.tar.gz
tar -xzvf 7_9.tar.gz训练数据集这是一个蛋白质-配体数据集,来源于 MOAD 数据集。每个文件压缩 (tar.gz) 的名字表示以相同初始 ID 开头的 PDB ID 集合,例如,1.tar.gz 包含所有以 1 开头的 PDB ID。作者以 ./data/1/1a0f 文件夹中的内容为例,介绍每个 PDB ID 对应的蛋白-配体复合物处理后的文件。./data/1/1a0f 文件夹中的内容如下:
.
|-- GTS_A_203.json
|-- GTS_A_203.sdf
|-- GTS_B_203.json
|-- GTS_B_203.sdf
`-- protein.pdb
0 directories, 5 files作者对 MOAD 数据集做了数据预处理,对于每个子文件夹,protein.pdb 都是准备好的蛋白质结构文件。这些文件已经过处理,其中包含添加正确的氢原子、质子化信息以及缺失的重原子和残基。具有相同名称的 JSON 和 SDF 文件记录一个配体,其命名格式为:配体名字_配体所在的链_残基编号。SDF 文件为配体原子坐标提供了正确的键序信息。JSON 文件包含有关配体重原子中心坐标的信息和配体周围 10 埃的盒子,可以直接用作分子对接中的网格参数。例如 GTS_A_203.json 的内容如下所示。包含 GTS 的重原子坐标中心坐标以及三个维度上的对接盒子尺寸。
{
"center_x": 37.08304214477539,
"center_y": -34.803218841552734,
"center_z": 66.12195587158203,
"size_x": 19.91899871826172,
"size_y": 18.468000411987305,
"size_z": 17.5469970703125
}项目提供训练好的模型权重文件 unimol_docking_v2_240517.pt ,下载链接为:https://www.dropbox.com/scl/fi/sfhrtx1tjprce18wbvmdr/unimol_docking_v2_240517.pt?rlkey=5zg7bh150kcinalrqdhzmyyoo&st=n6j0nt6c&dl=0。创建 ./weights 文件夹,把下载的模型权重文件上传到该文件夹。
3.2 安装环境
复制代码项目:
git clone https://github.com/deepmodeling/Uni-Mol.git下载的项目是 Uni-Mol,项目目录如下。其中 unimol_docking_v2 是此次测评的 Uni-Mol Docking V2 对接模型的文件夹,也是本次测评的目录。
.
├── LICENSE
├── README.md
├── docs
├── unimol
├── unimol2
├── unimol_docking_v2
├── unimol_plus
└── unimol_tools
6 directories, 2 files创建 Unimol_DockV2 运行环境并激活,命令如下:
conda create -n Unimol_DockV2 python=3.9
conda activate Unimol_DockV2项目运行环境需要安装 Uni-Core 项目的环境依赖,复制 Uni-Core 项目:
git clone https://github.com/dptech-corp/Uni-Core.git安装 torch 等环境依赖库:
pip3 install torch torchvision torchaudio --index-url https://download.pytorch.org/whl/cu118
安装 Uni-Core:
cd ./Uni-Core
pip install .安装其他依赖库:
pip install biopandas tqdm scikit-learn
pip install rdkit-pypi==2022.9.3 -i https://pypi.tuna.tsinghua.edu.cn/simple/ --trusted-host pypi.tuna.tsinghua.edu.cn
conda install rdkit==2022.9.33.3 对接构象生成案例测试
项目提供推断对接构象的脚本 ./interface/demo.py 运行脚本可以通过命令行的形式,添加相应的参数控制对接过程。下面对主要的参数进行简单介绍。
optional arguments:
-h, --help show this help message and exit
--model-dir MODEL_DIR
dir of the model
--input-protein INPUT_PROTEIN
path of the protein pdb file
--input-ligand INPUT_LIGAND
path of the ligand sdf file
--input-batch-file INPUT_BATCH_FILE
path of thr input file in batch mode, one line for each ligand, each line contains the input ligand path, the input docking grid path, and the output
ligand name
--input-docking-grid INPUT_DOCKING_GRID
name of the docking grid json file
--output-ligand-name OUTPUT_LIGAND_NAME
name of the ligand sdf file
--output-ligand-dir OUTPUT_LIGAND_DIR
name of the ligand sdf dir
--mode {single,batch_one2one,batch_one2many}
docking running mode, single and batch, batch_one2one represents batch_protein_to_single_ligand, batch_one2many represents
batch_protein_to_many_ligands,
--batch-size BATCH_SIZE
--nthreads NTHREADS num of threads for data preprocessing
--conf-size CONF_SIZE
number of conformers generated with each molecule
--cluster whether preform conformer clustering when data preprocess
--use_current_ligand_conf
--steric-clash-fix Whether to perform steric clash fix on Unimol docking results
其中,--model-dir 用来指定训练好的模型参数文件,比如如: ./weights/unimol_docking_v2_240517.pt 。--input-protein 指定输入蛋白的 pdb 格式的结构文件,如果是批量预测对接构象,可以传入一个蛋白结构文件的列表。--input-ligand 指定输入要对接的化合物 sdf 格式的结构文件,如果是批量预测对接构象,可以传入一个化合物结构文件的列表。--input-docking-grid 指定一个 JSON 格式的文件,其中包含对接的中心坐标和对接盒子的大小,如果是批量预测对接构象,可以传入一个 JSON 文件的列表。--output-ligand-name 指定输出预测对接构象配体的文件名,如果是批量预测对接构象,可以传入一个文件名的列表。--output-ligand-dir 指定保存于的对接构象的文件夹。--mode 用来指定运行的模式,可选(1)single ,表示一个蛋白结构和化合物结构作为输入。(2)batch_one2one,表示批量蛋白和批量化合物作为输入,蛋白结构和化合物结构数量一致,一一对应预测对接构象。(3)batch_one2many,表示一个蛋白和批量化合物作为输入,预测全部化合物在该蛋白口袋中的对接构象。--steric-clash-fix 用来指定是否对对接结果执行空间冲突修复。
3.3.1 内置案例
项目提供一个内置的测试案例来评估 Uni-Mol Docking V2 的表现。内置案例是 1A30 蛋白,配体在口袋中的构象如下。需要指出的是,配体并不是 1A30 蛋白晶体结构中的,而是作者另外提供的。
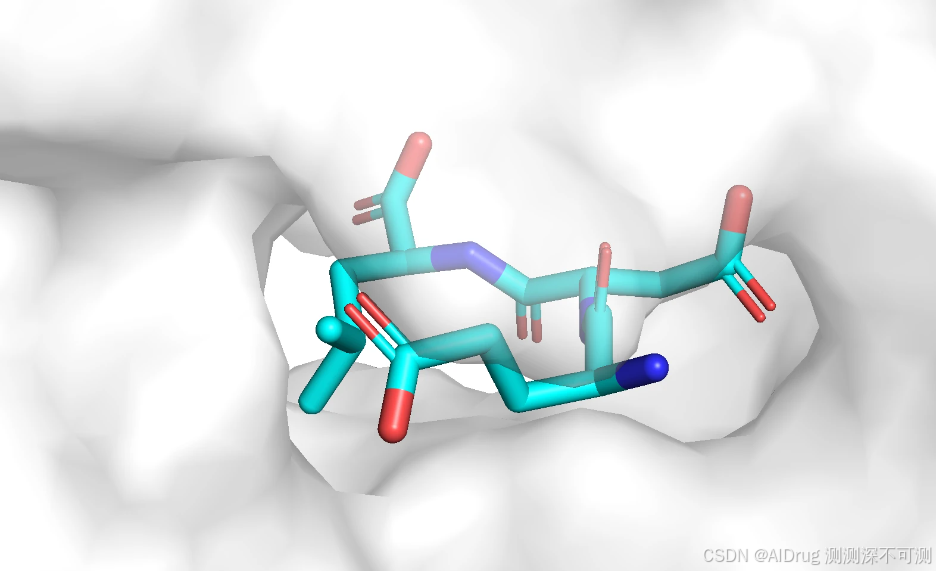
配体的 2D 结构如下所示:
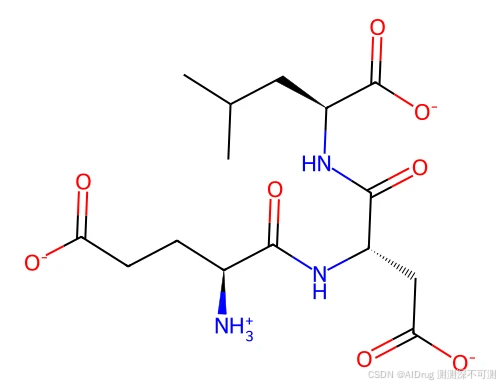
处理好的数据在 ./example_data 文件夹中,目录如下:
.
|-- dict_mol.txt
|-- dict_pkt.txt
|-- docking_grid.json
|-- ligand.sdf
`-- protein.pdb
0 directories, 5 files其中,ligand.sdf 和 protein.pdb 是模型输入的化合物和蛋白结构文件。docking_grid.json 中包含了化合物的中心坐标和对接盒子的尺寸信息。
激活 Unimol_DockV2 环境。
conda activate Unimol_DockV2
通过下面命令指定内置案例的蛋白和配体结构文件、对接盒子信息、以及输出路径,执行目录在 ./。
python ./interface/demo.py \
--model-dir ./weights/unimol_docking_v2_240517.pt \
--input-protein ./example_data/protein.pdb \
--input-ligand ./example_data/ligand.sdf \
--input-docking-grid ./example_data/docking_grid.json \
--output-ligand-name pose_pred \
--output-ligand-dir ./example_data/output \
--steric-clash-fix \
--mode single运行输出如下:
Namespace(model_dir='./weights/unimol_docking_v2_240517.pt', input_protein='./example_data/protein.pdb', input_ligand='./example_data/ligand.sdf', input_batch_file='input_batch.csv', input_docking_grid='./example_data/docking_grid.json', output_ligand_name='pose_pred', output_ligand_dir='./example_data/output', mode='single', batch_size=4, nthreads=8, conf_size=10, cluster=False, use_current_ligand_conf=False, steric_clash_fix=True)
Start preprocessing data...
Number of ligands: 1
1it [00:01, 1.42s/it]
Total num: 1, Success: 1, Failed: 0
Done!
fused_multi_tensor is not installed corrected
fused_rounding is not installed corrected
fused_layer_norm is not installed corrected
fused_softmax is not installed corrected
2024-12-04 12:36:36 | INFO | unimol.inference | loading model(s) from ./weights/unimol_docking_v2_240517.pt
/workspace/anaconda3/envs/Unimol_DockV2/lib/python3.9/site-packages/unicore/checkpoint_utils.py:251: FutureWarning: You are using `torch.load` with `weights_only=False` (the current default value), which uses the default pickle module implicitly. It is possible to construct malicious pickle data which will execute arbitrary code during unpickling (See https://github.com/pytorch/pytorch/blob/main/SECURITY.md#untrusted-models for more details). In a future release, the default value for `weights_only` will be flipped to `True`. This limits the functions that could be executed during unpickling. Arbitrary objects will no longer be allowed to be loaded via this mode unless they are explicitly allowlisted by the user via `torch.serialization.add_safe_globals`. We recommend you start setting `weights_only=True` for any use case where you don't have full control of the loaded file. Please open an issue on GitHub for any issues related to this experimental feature.
state = torch.load(f, map_location=torch.device("cpu"))
2024-12-04 12:36:36 | INFO | unimol.tasks.docking_pose_v2 | ligand dictionary: 30 types
2024-12-04 12:36:36 | INFO | unimol.tasks.docking_pose_v2 | pocket dictionary: 9 types
2024-12-04 12:36:37 | INFO | unimol.inference | Namespace(no_progress_bar=False, log_interval=50, log_format='simple', tensorboard_logdir='', wandb_project='', wandb_name='', seed=1, cpu=False, fp16=True, bf16=False, bf16_sr=False, allreduce_fp32_grad=False, fp16_no_flatten_grads=False, fp16_init_scale=4, fp16_scale_window=256, fp16_scale_tolerance=0.0, min_loss_scale=0.0001, threshold_loss_scale=None, user_dir='/workspace/XXXX/projects/unimol_docking_v2/unimol', empty_cache_freq=0, all_gather_list_size=16384, suppress_crashes=False, profile=False, ema_decay=-1.0, validate_with_ema=False, loss='docking_pose_v2', optimizer='adam', lr_scheduler='fixed', task='docking_pose_v2', num_workers=8, skip_invalid_size_inputs_valid_test=False, batch_size=4, required_batch_size_multiple=1, data_buffer_size=10, train_subset='train', valid_subset='pose_pred', validate_interval=1, validate_interval_updates=0, validate_after_updates=0, fixed_validation_seed=None, disable_validation=False, batch_size_valid=4, max_valid_steps=None, curriculum=0, distributed_world_size=1, distributed_rank=0, distributed_backend='nccl', distributed_init_method=None, distributed_port=-1, device_id=0, distributed_no_spawn=False, ddp_backend='c10d', bucket_cap_mb=25, fix_batches_to_gpus=False, find_unused_parameters=False, fast_stat_sync=False, broadcast_buffers=False, nprocs_per_node=1, path='./weights/unimol_docking_v2_240517.pt', quiet=False, model_overrides='{}', results_path='/workspace/XXXX/projects/unimol_docking_v2/example_data/output', arch='docking_pose_v2', recycling=4, data='/workspace/XXXX/projects/unimol_docking_v2/example_data/output', finetune_mol_model=None, finetune_pocket_model=None, conf_size=10, dist_threshold=8.0, max_pocket_atoms=256, adam_betas='(0.9, 0.999)', adam_eps=1e-08, weight_decay=0.0, force_anneal=None, lr_shrink=0.1, warmup_updates=0, no_seed_provided=False, mol=Namespace(encoder_layers=15, encoder_embed_dim=512, encoder_ffn_embed_dim=2048, encoder_attention_heads=64, dropout=0.1, emb_dropout=0.1, attention_dropout=0.1, activation_dropout=0.0, pooler_dropout=0.0, max_seq_len=512, activation_fn='gelu', pooler_activation_fn='tanh', post_ln=False, masked_token_loss=-1.0, masked_coord_loss=-1.0, masked_dist_loss=-1.0, x_norm_loss=-1.0, delta_pair_repr_norm_loss=-1.0), pocket=Namespace(encoder_layers=15, encoder_embed_dim=512, encoder_ffn_embed_dim=2048, encoder_attention_heads=64, dropout=0.1, emb_dropout=0.1, attention_dropout=0.1, activation_dropout=0.0, pooler_dropout=0.0, max_seq_len=512, activation_fn='gelu', pooler_activation_fn='tanh', post_ln=False, masked_token_loss=-1.0, masked_coord_loss=-1.0, masked_dist_loss=-1.0, x_norm_loss=-1.0, delta_pair_repr_norm_loss=-1.0), encoder_layers=15, encoder_embed_dim=512, encoder_ffn_embed_dim=2048, encoder_attention_heads=64, dropout=0.1, emb_dropout=0.1, attention_dropout=0.1, activation_dropout=0.0, pooler_dropout=0.0, max_seq_len=512, activation_fn='gelu', pooler_activation_fn='tanh', post_ln=False, masked_token_loss=-1.0, masked_coord_loss=-1.0, masked_dist_loss=-1.0, x_norm_loss=-1.0, delta_pair_repr_norm_loss=-1.0, distributed_num_procs=1)
2024-12-04 12:36:37 | INFO | unicore.tasks.unicore_task | get EpochBatchIterator for epoch 1
2024-12-04 12:36:40 | INFO | unimol.inference | Done inference!
Start converting model predictions into sdf files...
100%|████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████| 1/1 [00:00<00:00, 1498.50it/s]
Done!
0%| | 0/1 [00:00<?, ?it/s]./example_data/output/pose_pred.sdf-CC(C)C[C@H](NC(=O)[C@H](CC(=O)[O-])NC(=O)[C@@H]([NH3+])CCC(=O)[O-])C(=O)[O-]-RMSD:6.2968
100%|██████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████| 1/1 [00:09<00:00, 9.35s/it]
output ligand path:
['./example_data/output/pose_pred.sdf']
total time: 19.374167442321777 sec.
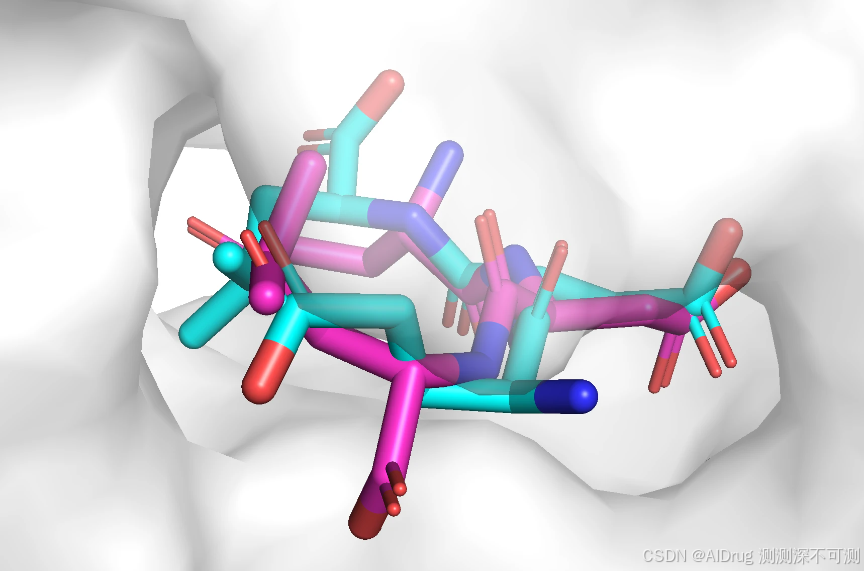
All processes done!对接运行大约耗时 20 秒,显存占用约为 1.5 GB,输出结果保存在 ./example_data/output 文件夹中。预测的对接构象为 ./example_data/output/pose_pred.sdf 。对接构象(紫红色)在口袋中的位置如下,蓝色表示输入的配体。由于模型输入限制了对接盒子的大小以及对接中心坐标,所以生成构象和输入构象的在口袋中的位置基本一致,和口袋残基没有空间冲突问题。
3.3.2 自定义案例(ABL1)
,我们设置了 ABL1 体系。6HD6_allo_protein.pdb,属于 Holo 结构,在此结构中,存在两个小分子,一个底物小分子抑制剂 Imatinib,另一个是变构小分子 Compound 6。除了底物分子 Imatinib 和 变构分子 Compound 6,我们还找到了如下分子。其中, Compound 5 和 Compound 7 均属于变构分子,且活性与 Compound 6有明显的区别。Compound N 则是来源于非激酶体系的变构小分子,是肯定不能结合在底物或者变构位点的。关于 ABL1 测试体系的详细介绍,可以参考前面介绍 FlexPose 的文章。
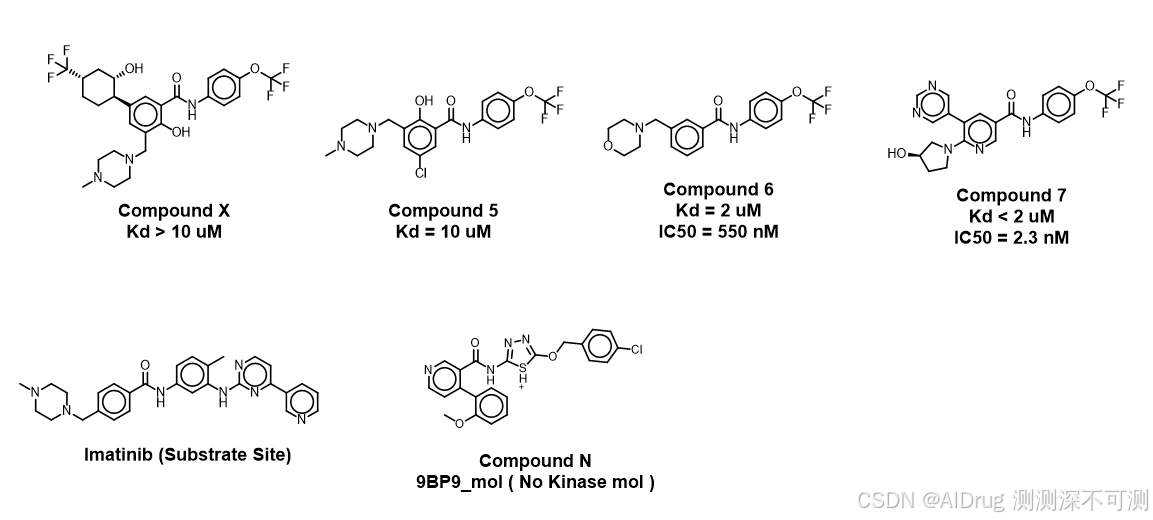
注:在上述分子中,Imatinib 和 Compound X 没有进行测试。
将所有的测试i体系文件放置在 ./example_ABL1 文件夹内,Protein 中保存蛋白 PDB 文件,compound 文件夹中保存小分子的 3D 文件。
.
|-- bust_output
|-- compound
|-- docking_grid.json
|-- input_batch_one2many.csv
|-- output
|-- pred_bust_results.csv
`-- proteion
4 directories, 3 files
(Unimol_DockV2) wufeil@7ab82433c90d655e:/workspace/XXXX/projects/unimol_docking_v2/example_ABL1$ tree -L 2
|-- compound
| |-- compound_5.sdf
| |-- compound_6.sdf
| |-- compound_7.sdf
| `-- compound_N.sdf
`-- proteion
`-- 6HD6_allo_protein.pdb
2 directories, 5 files
化合物 6 是晶体结构,所以我们根据它来确定模型输入需要的对接中心坐标和对接盒子大小。通过 PyMol 打开化合物 6 ,命名为 compound_6,使用 get_extent 获取配体三个坐标维度上的最大值和最小值,命令如下:
get_extent compound_6返回结果如下:
cmd.extent: min: [ 17.155, 37.536, 49.132]
cmd.extent: max: [ 27.164, 48.675, 59.914]根据返回结果创建 ./example_ABL1/docking_grid.json 来指定对接中心坐标和对接盒子的尺寸,具体内容如下:
{
"center_x": 22.1595,
"center_y": 43.1055,
"center_z": 54.523,
"size_x": 20.009,
"size_y": 21.139,
"size_z": 20.782
}创建 ./example_ABL1/compound 把要对接的化合物结构文件保存到该文件夹,目录如下:
.
|-- compound_5.sdf
|-- compound_6.sdf
|-- compound_7.sdf
`-- compound_N.sdf
0 directories, 4 files我们要把4 个分子都对接到同一个蛋白口袋中,属于 batch_one2many 运行模式。需要把模型的输入信息以一个 CSV 表格的形式传入。创建 ./prepare_ABL1.ipynb 生成包含输入信息的 CSV 格式表格,代码如下:
import os
import pandas as pd
cpd_name = os.listdir('./example_ABL1/compound')
input_ligand = ['./example_ABL1/compound/' + cpd for cpd in os.listdir('./example_ABL1/compound')]
input_docking_grid = ['./example_ABL1/docking_grid.json'] * len(input_ligand)
output_ligand_name = [x.split('.')[0] + '_pred' for x in cpd_name]
df = pd.DataFrame({'input_ligand': input_ligand, 'input_docking_grid': input_docking_grid, 'output_ligand_name': output_ligand_name})
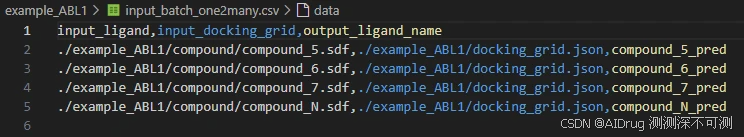
df.to_csv('./example_ABL1/input_batch_one2many.csv', index=False)运行结束,生成包含输入信息的 CSV 表格,内容如下:
通过下面命令对接测试案例,指定对接构象输出文件夹为 ./example_ABL1/output。
python ./interface/demo.py \
--model-dir ./weights/unimol_docking_v2_240517.pt \
--input-protein ./example_ABL1/proteion/6HD6_allo_protein.pdb \
--input-batch-file ./example_ABL1/input_batch_one2many.csv \
--output-ligand-dir ./example_ABL1/output \
--steric-clash-fix \
--mode batch_one2many运行输出如下:
Namespace(model_dir='./weights/unimol_docking_v2_240517.pt', input_protein='./example_ABL1/proteion/6HD6_allo_protein.pdb', input_ligand='ligand.sdf', input_batch_file='./example_ABL1/input_batch_one2many.csv', input_docking_grid='docking_grid.json', output_ligand_name='ligand_predict', output_ligand_dir='./example_ABL1/output_test', mode='batch_one2many', batch_size=4, nthreads=8, conf_size=10, cluster=False, use_current_ligand_conf=False, steric_clash_fix=True)
Start preprocessing data...
Number of ligands: 4
4it [00:01, 2.83it/s]
Total num: 4, Success: 4, Failed: 0
Done!
fused_multi_tensor is not installed corrected
fused_rounding is not installed corrected
fused_layer_norm is not installed corrected
fused_softmax is not installed corrected
2024-12-04 11:21:40 | INFO | unimol.inference | loading model(s) from ./weights/unimol_docking_v2_240517.pt
/workspace/anaconda3/envs/Unimol_DockV2/lib/python3.9/site-packages/unicore/checkpoint_utils.py:251: FutureWarning: You are using `torch.load` with `weights_only=False` (the current default value), which uses the default pickle module implicitly. It is possible to construct malicious pickle data which will execute arbitrary code during unpickling (See https://github.com/pytorch/pytorch/blob/main/SECURITY.md#untrusted-models for more details). In a future release, the default value for `weights_only` will be flipped to `True`. This limits the functions that could be executed during unpickling. Arbitrary objects will no longer be allowed to be loaded via this mode unless they are explicitly allowlisted by the user via `torch.serialization.add_safe_globals`. We recommend you start setting `weights_only=True` for any use case where you don't have full control of the loaded file. Please open an issue on GitHub for any issues related to this experimental feature.
state = torch.load(f, map_location=torch.device("cpu"))
2024-12-04 11:21:40 | INFO | unimol.tasks.docking_pose_v2 | ligand dictionary: 30 types
2024-12-04 11:21:40 | INFO | unimol.tasks.docking_pose_v2 | pocket dictionary: 9 types
2024-12-04 11:21:42 | INFO | unimol.inference | Namespace(no_progress_bar=False, log_interval=50, log_format='simple', tensorboard_logdir='', wandb_project='', wandb_name='', seed=1, cpu=False, fp16=True, bf16=False, bf16_sr=False, allreduce_fp32_grad=False, fp16_no_flatten_grads=False, fp16_init_scale=4, fp16_scale_window=256, fp16_scale_tolerance=0.0, min_loss_scale=0.0001, threshold_loss_scale=None, user_dir='/workspace/XXXX/projects/unimol_docking_v2/unimol', empty_cache_freq=0, all_gather_list_size=16384, suppress_crashes=False, profile=False, ema_decay=-1.0, validate_with_ema=False, loss='docking_pose_v2', optimizer='adam', lr_scheduler='fixed', task='docking_pose_v2', num_workers=8, skip_invalid_size_inputs_valid_test=False, batch_size=4, required_batch_size_multiple=1, data_buffer_size=10, train_subset='train', valid_subset='batch_data', validate_interval=1, validate_interval_updates=0, validate_after_updates=0, fixed_validation_seed=None, disable_validation=False, batch_size_valid=4, max_valid_steps=None, curriculum=0, distributed_world_size=1, distributed_rank=0, distributed_backend='nccl', distributed_init_method=None, distributed_port=-1, device_id=0, distributed_no_spawn=False, ddp_backend='c10d', bucket_cap_mb=25, fix_batches_to_gpus=False, find_unused_parameters=False, fast_stat_sync=False, broadcast_buffers=False, nprocs_per_node=1, path='./weights/unimol_docking_v2_240517.pt', quiet=False, model_overrides='{}', results_path='/workspace/XXXX/projects/unimol_docking_v2/example_ABL1/output_test', arch='docking_pose_v2', recycling=4, data='/workspace/XXXX/projects/unimol_docking_v2/example_ABL1/output_test', finetune_mol_model=None, finetune_pocket_model=None, conf_size=10, dist_threshold=8.0, max_pocket_atoms=256, adam_betas='(0.9, 0.999)', adam_eps=1e-08, weight_decay=0.0, force_anneal=None, lr_shrink=0.1, warmup_updates=0, no_seed_provided=False, mol=Namespace(encoder_layers=15, encoder_embed_dim=512, encoder_ffn_embed_dim=2048, encoder_attention_heads=64, dropout=0.1, emb_dropout=0.1, attention_dropout=0.1, activation_dropout=0.0, pooler_dropout=0.0, max_seq_len=512, activation_fn='gelu', pooler_activation_fn='tanh', post_ln=False, masked_token_loss=-1.0, masked_coord_loss=-1.0, masked_dist_loss=-1.0, x_norm_loss=-1.0, delta_pair_repr_norm_loss=-1.0), pocket=Namespace(encoder_layers=15, encoder_embed_dim=512, encoder_ffn_embed_dim=2048, encoder_attention_heads=64, dropout=0.1, emb_dropout=0.1, attention_dropout=0.1, activation_dropout=0.0, pooler_dropout=0.0, max_seq_len=512, activation_fn='gelu', pooler_activation_fn='tanh', post_ln=False, masked_token_loss=-1.0, masked_coord_loss=-1.0, masked_dist_loss=-1.0, x_norm_loss=-1.0, delta_pair_repr_norm_loss=-1.0), encoder_layers=15, encoder_embed_dim=512, encoder_ffn_embed_dim=2048, encoder_attention_heads=64, dropout=0.1, emb_dropout=0.1, attention_dropout=0.1, activation_dropout=0.0, pooler_dropout=0.0, max_seq_len=512, activation_fn='gelu', pooler_activation_fn='tanh', post_ln=False, masked_token_loss=-1.0, masked_coord_loss=-1.0, masked_dist_loss=-1.0, x_norm_loss=-1.0, delta_pair_repr_norm_loss=-1.0, distributed_num_procs=1)
2024-12-04 11:21:42 | INFO | unicore.tasks.unicore_task | get EpochBatchIterator for epoch 1
2024-12-04 11:21:53 | INFO | unimol.inference | Done inference!
Start converting model predictions into sdf files...
100%|████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████| 4/4 [00:00<00:00, 1947.22it/s]
Done!
0%| | 0/4 [00:00<?, ?it/s]./example_ABL1/output_test/compound_5_pred.sdf-CN1CCN(Cc2cc(Cl)cc(C(=O)Nc3ccc(OC(F)(F)F)cc3)c2O)CC1-RMSD:2.1472
25%|████████████████████████████████████████████████████▌ | 1/4 [00:07<00:21, 7.16s/it]./example_ABL1/output_test/compound_6_pred.sdf-O=C(Nc1ccc(OC(F)(F)F)cc1)c1cccc(CN2CCOCC2)c1-RMSD:0.9324
50%|█████████████████████████████████████████████████████████████████████████████████████████████████████████ | 2/4 [00:07<00:06, 3.30s/it][11:22:03] Can't kekulize mol. Unkekulized atoms: 0 1 2 18 19
Traceback (most recent call last):
File "/workspace/XXXX/projects/unimol_docking_v2/unimol/scripts/6tsr.py", line 603, in <module>
single_refine(
File "/workspace/XXXX/projects/unimol_docking_v2/unimol/scripts/6tsr.py", line 561, in single_refine
label_lig = Chem.RemoveHs(label_lig)
rdkit.Chem.rdchem.KekulizeException: Can't kekulize mol. Unkekulized atoms: 0 1 2 18 19
./example_ABL1/output_test/compound_7_pred.sdf-O=C(Nc1ccc(OC(F)(F)F)cc1)c1cnc(N2CCC(O)C2)c(-c2cncnc2)c1-RMSD:1.4946
100%|██████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████| 4/4 [00:08<00:00, 2.06s/it]
output ligands path:
['./example_ABL1/output_test/compound_5_pred.sdf', './example_ABL1/output_test/compound_6_pred.sdf', './example_ABL1/output_test/compound_7_pred.sdf', './example_ABL1/output_test/compound_N_pred.sdf']
Average time: 6.683359622955322 sec.
Total time: 26.73343849182129 sec.
All processes done!对接运行大约耗时 30 秒,显存占用约为 2 GB,输出结果保存在 ./example_ABL1/output 文件夹中,文件目录如下,其中的四个 sdf 格式文件分别对应预测的构象。
.
|-- batch_data.lmdb
|-- batch_data.pkl
|-- compound_5_pred.sdf
|-- compound_6_pred.sdf
|-- compound_7_pred.sdf
|-- compound_N_pred.sdf
|-- dict_mol.txt
`-- dict_pkt.txt
0 directories, 8 files生成构象在口袋中的 pse 文件如下(compound_6_ligand 是参考的晶体结构)。由于指定了对接中心坐标和对接盒子,所以所有化合物的对接构象在口袋中的朝向和位置基本一致。对接构象在口袋中的位置如下所示:
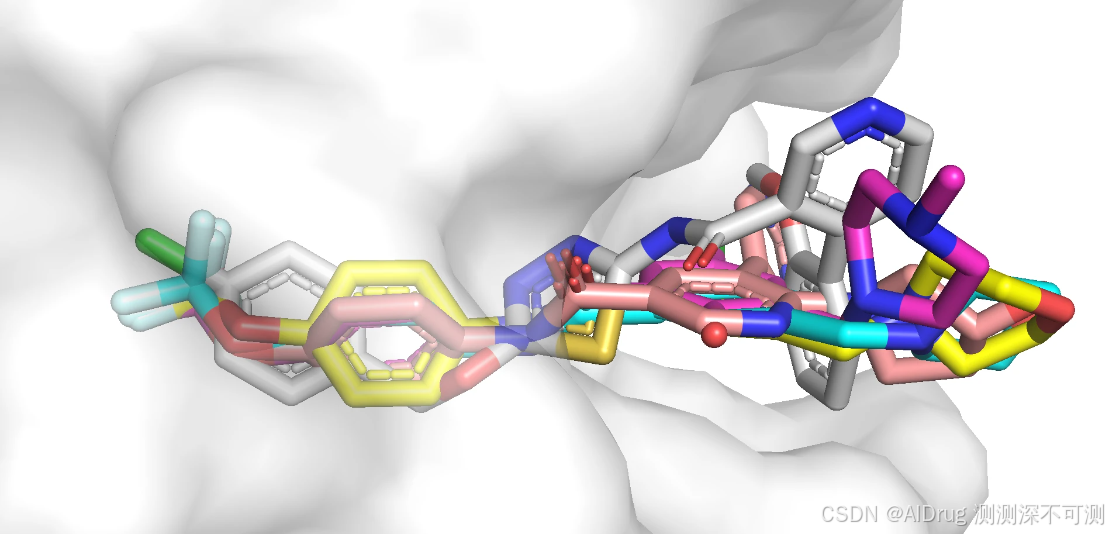
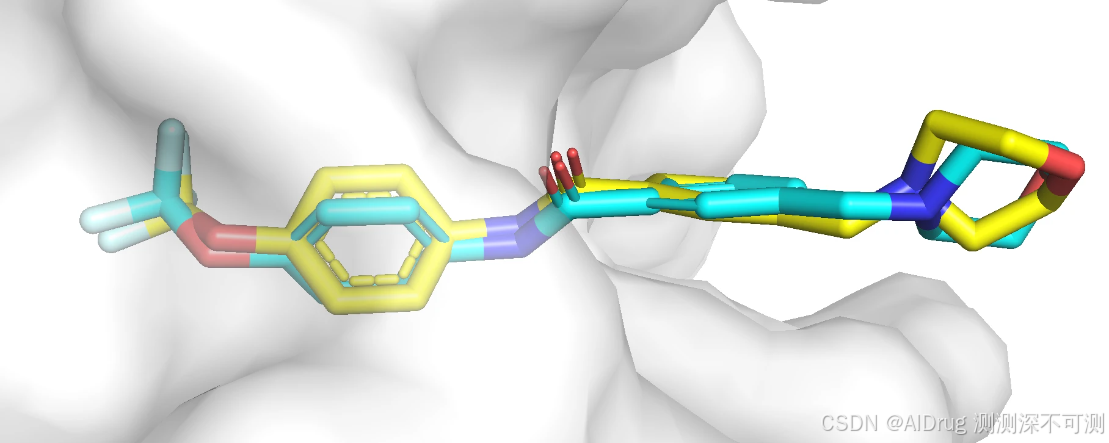
下面展示化合物 6 的晶体结构(蓝色)和对接构象(黄色),对结构象和晶体结构的位置和朝向是一致的,两个构象基本一样,预测效果良好。
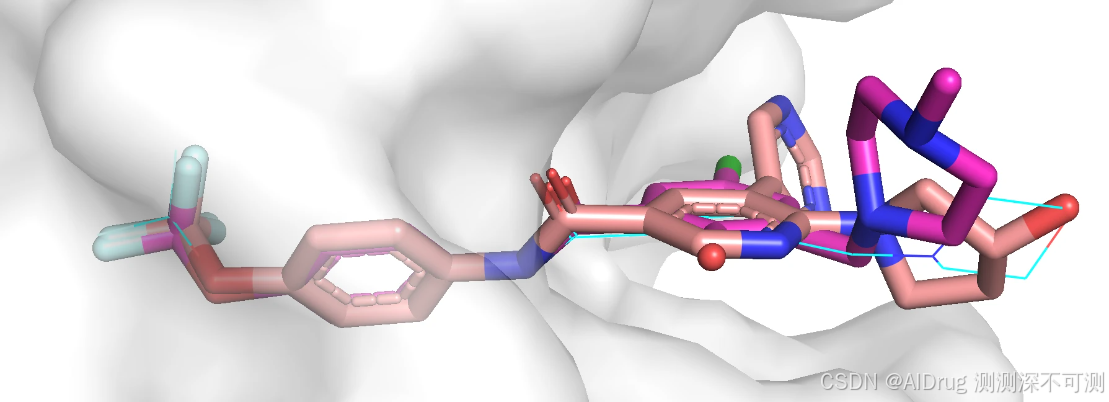
化合物 5 (紫红色)和 7 (浅粉色)的对接构象、化合物 6 的晶体结构(蓝色)如下所示,口袋中相同的片段基本重合,溶剂区的片段有部分摆动。
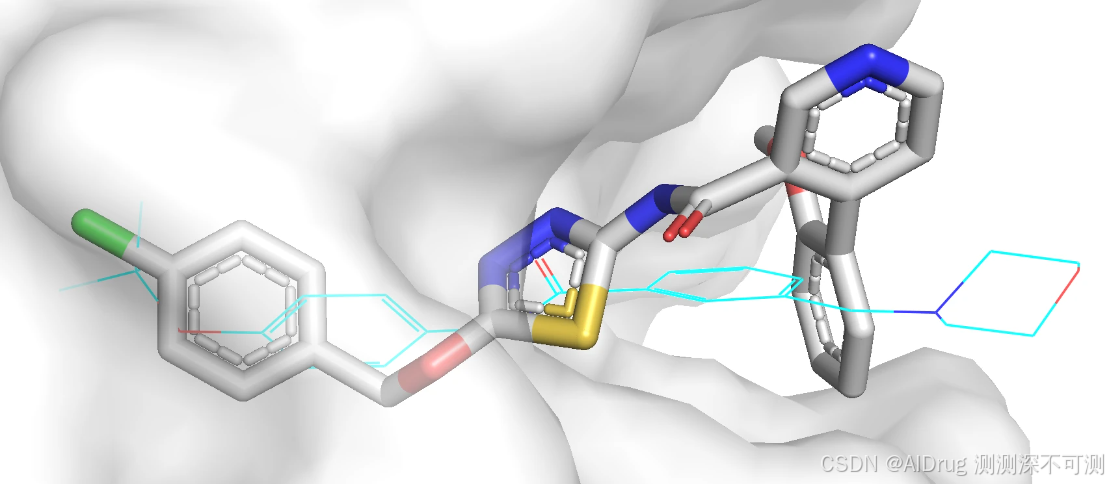
化合物 N (灰色)和化合物 6 (蓝色)的 2D 结构存在较大差异,所以构象也是差异最大的。
使用 ./pb_screen_mols.py 脚本,通过 PoseBusters 检查分子的对接构象在化学和物理方面的有效性。创建 ./ABL1_output/cpd_dock 文件夹,把预测的构象复制到该文件夹。注:我们使用 RDKit 查看对接构象时发现,compound_N 无法被读取,所以其生成的构象并不是有效分子,下面不用再检查了。
通过 PoseBusters 评价预测的分子构象:激活 PoseBusters 环境:
conda activate posebusters过下面命令运行 PoseBusters 检查构象:
python ./pb_screen_mols.py \
--input_molecule_dir ./example_ABL1/output/cpd_dock \
--reference_protein_path ./example_ABL1/proteion/6HD6_allo_protein.pdb \
--output_molecule_dir ./example_ABL1/bust_output \
--bust_results_filepath ./example_ABL1/pred_bust_results.csv其中,--input_molecule_dir 指定生成分子所在的文件夹,即 ./example_ABL1/output/cpd_dock 文件夹。--reference_protein_path 指定不包含参考配体的蛋白结构文件(./example_ABL1/proteion/6HD6_allo_protein.pdb)。--output_molecule_dir 指定评价过程中生成分子产生的缓存结构的文件夹。--bust_results_filepath 保存每个生成分子在各项指标的通过情况,这里保存为 bust_results.csv 表格。我们在已有的 PoseBusters 环境中运行 PoseBusters 评价的脚本。
模型输出:
Found ['./example_ABL1/output/cpd_dock/compound_5_pred.sdf', './example_ABL1/output/cpd_dock/compound_6_pred.sdf', './example_ABL1/output/cpd_dock/compound_7_pred.sdf'] SDF files in ./example_ABL1/output/cpd_dock.
PoseBusters results for input molecule directory ./example_ABL1/output/cpd_dock saved to ./example_ABL1/pred_bust_results.csv.
PoseBuster-valid molecules saved to ./example_ABL1/bust_output.评价的分子顺序列表是 ['./example_ABL1/output/cpd_dock/compound_5_pred.sdf', './example_ABL1/output/cpd_dock/compound_6_pred.sdf', './example_ABL1/output/cpd_dock/compound_7_pred.sdf']
对应的检查结果如下(./example_ABL1/pred_bust_results.csv):
mol_pred_loaded,mol_cond_loaded,sanitization,all_atoms_connected,bond_lengths,bond_angles,internal_steric_clash,aromatic_ring_flatness,double_bond_flatness,internal_energy,protein-ligand_maximum_distance,minimum_distance_to_protein,minimum_distance_to_organic_cofactors,minimum_distance_to_inorganic_cofactors,minimum_distance_to_waters,volume_overlap_with_protein,volume_overlap_with_organic_cofactors,volume_overlap_with_inorganic_cofactors,volume_overlap_with_waters
True,True,True,True,True,True,True,True,True,True,True,True,True,True,True,True,True,True,True
True,True,True,True,True,True,True,True,True,True,True,True,True,True,True,True,True,True,True
True,True,True,True,True,True,True,True,True,True,True,True,True,True,True,True,True,True,True从返回的评估结果来看,化合物 5、6 和 7 的预测构象通过了全部检查。表明了 Uni-Mol Docking V2 模型的良好表现。
通过内置案例和自定义案例的测试结果,我们发现 Uni-Mol Docking V2 预测模型的表现良好。因为根据参考化合物指定了对接盒子,限制了化合物的构象空间,所以没有出现和蛋白质口袋残基空间冲突的问题,生成分子的构象也比较合理。
compound_6.sdf 是晶体结构中的化合物构象,我们把它作为参考分子,计算对接构象的 RMSD 。对接构象和化合物 6 的 RMSD 的结果如下(括号内的元组分别对应 RMSD 和匹配的最大原子数目):
RMSD(compound_5_pred.sdf): (0.54, 26)
RMSD(compound_6_pred.sdf): (0.42, 27)
RMSD(compound_7_pred.sdf): (0.53, 27)以下是 Uni-Mol Docking V2 在变构口袋上预测的各个小分子的最大公共子片段 RMSD,PoseBusters 检查通过情况以及与原本的 Kd、IC50,具体见下表:
| IC50 (uM) | Kd (uM) | RMSD | PoseBusters check | |
| Compound 5 | / | 10 | 0.54 | True |
| Compound 6 | 550 | 2 | 0.42 | True |
| Compound 7 | 0.0023 | <2 | 0.53 | True |
| Compound N | False | False | / | False |
3.3.4 训练模型
Uni-Mol Docking V2 是基于 Uni-Mol 微调得到的,可以从 Uni-Mol 项目中获取预训练好的蛋白和小分子的模型权重文件,链接是:https://github.com/deepmodeling/Uni-Mol/tree/main/unimol,网页部分如下所示:
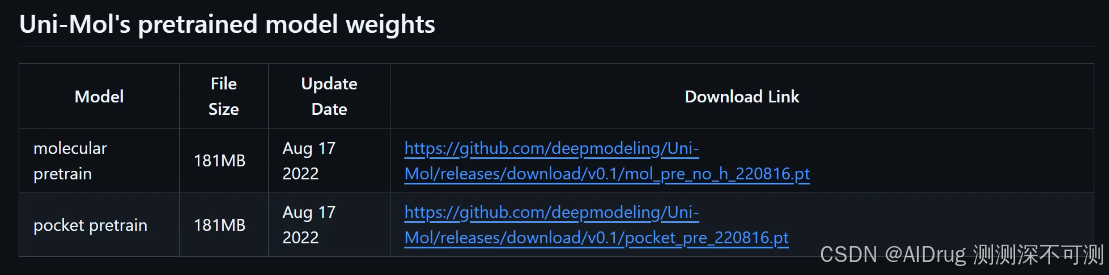
预训练的小分子权重文件链接是:https://github.com/deepmodeling/Uni-Mol/releases/download/v0.1/mol_pre_no_h_220816.pt。预训练的蛋白质口袋权重文件链接是:https://github.com/deepmodeling/Uni-Mol/releases/download/v0.1/pocket_pre_220816.pt 。下载后上传到 ./weights 文件夹中。
下载划分好的训练集、验证集和测试集数据,连接为:https://bioos-hermite-beijing.tos-cn-beijing.volces.com/unimol_data/finetune/protein_ligand_binding_pose_prediction.tar.gz ,下载后解压后把划分好的数据集 LMDB 文件上传到 .data/ 文件夹中。训练还需要 dict_mol.txt 和 dict_pkt.txt 来指定化合物和蛋白口袋允许的特殊标记和元素类型等,这两个文件从 ./example_data 中复制到 ./data 中,以便程序读取。比如 dict_pkt.txt 的内容如下所示,[PAD] 表示填充(padding)符号,用于保持输入序列的长度一致。[CLS]:表示分类符号(classification token),用于表示整个序列的表示。[SEP]:分隔符(separator),用于区分不同的句子或子任务。[UNK]:未知符号(unknown token),表示在模型中遇到未见过的词或符号。其余的是蛋白口袋中的化学元素符号。
[PAD]
[CLS]
[SEP]
[UNK]
C
N
O
S
H
至此,data 文件夹中已经准备好训练模型所需的文件,目录如下:
.
|-- dict_mol.txt
|-- dict_pkt.txt
|-- test.lmdb
|-- train.lmdb
`-- valid.lmdb
0 directories, 5 files项目提供 train.sh 可以方便的重新训练模型,我们修改该脚本内容以适应我们的机器,脚本的具体内容如下:
data_path="./data" # replace to your data path
save_dir="./save_model" # replace to your save path
n_gpu=1
MASTER_PORT=10086
finetune_mol_model="./weights/mol_pre_no_h_220816.pt"
finetune_pocket_model="./weights/pocket_pre_220816.pt"
lr=3e-5
batch_size=16
epoch=2
dropout=0.2
warmup=0.06
update_freq=1
dist_threshold=8.0
recycling=4
export NCCL_ASYNC_ERROR_HANDLING=1
export OMP_NUM_THREADS=1
# 使用 torchrun 启动分布式训练
python $(which unicore-train) --user-dir ./unimol $data_path --train-subset train --valid-subset valid \
--num-workers 1 --task docking_pose_v2 --loss docking_pose_v2 --arch docking_pose_v2 \
--optimizer adam --adam-betas '(0.9, 0.99)' --adam-eps 1e-6 --clip-norm 1.0 \
--lr-scheduler polynomial_decay --lr $lr --warmup-ratio $warmup --max-epoch $epoch --batch-size $batch_size \
--mol-pooler-dropout $dropout --pocket-pooler-dropout $dropout \
--update-freq $update_freq --seed 42 \
--fp16 --fp16-init-scale 4 --fp16-scale-window 256 \
--tensorboard-logdir $save_dir/tsb \
--log-interval 100 --log-format simple \
--validate-interval 1 --keep-last-epochs 10 \
--best-checkpoint-metric valid_loss --patience 2000 --all-gather-list-size 1024000 \
--finetune-mol-model $finetune_mol_model \
--finetune-pocket-model $finetune_pocket_model \
--dist-threshold $dist_threshold --recycling $recycling \
--save-dir $save_dir \
--find-unused-parameters \
--required-batch-size-multiple 1我们设置模型输出保存的文件夹为 ./save_model (save_dir)。batch_size 设置为 16,显存占用最大约为 24 GB。设置 epoch=2 ,测试训练两个 epoch 。
我们使用挂起的方式后台训练,把输出内容保存到 train.log 以供后续查看,训练命令如下:
nohup bash train.sh > train.log 2>&1 &训练规程记录到 train.log 中,我们测试训练了两个 epoch,耗时约 2 小时,显存占用最大约 24 GB 。每训练一个 epoch,就保存一个 checkpoint,保存目录为 ./save_model 。目录结构如下。除了保存每个 checkpoint 的模型,还会保存最后一个 epoch 和在验证集上表现最好的模型。
.
|-- checkpoint1.pt
|-- checkpoint2.pt
|-- checkpoint_best.pt
|-- checkpoint_last.pt
`-- tsb
1 directory, 4 files四、总结
最新版本的 UniMol Docking V2 在 PoseBusters 基准测试中确立了新的最前沿,超越了之前的版本,后者是截至 2023 年 11 月 22 日为止,表现最好的开源模型。在新的版本中,作者使用了更大的模型,以及对 MOAD 数据集进行了数据预处理。
测评结果显示,在我们自有的体系上 ABL 1 上,docking 出来的小分子构象与晶体构象还是非常相近的,同时小分子也能通过 PoseBuster 的小分子内,小分子-蛋白间的检测。小分子的构象也比较合理,比如:苯环共平面了,酰胺共平面了,整体构象基本OK。确实整体效果还不错,值得一试。(注:我们的测试体系已经出现在了他们的训练集中,在使用前,请使用自己的体系试一下)


















 876
876

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











