






写在前面,不推荐!不推荐!真有这个需求,用Homer更方便,Biomart还是用来注释就行了。
理由:①.Biomart里面Ensemble Regulation数据着实太少!人类转录因子结合位点数据相对而言多一点,而小鼠的转录因子数据,真的少得可怜。可能是因为经过验证过的数据才能放上来吧(纯属个人猜测)。不过组蛋白修饰结合位点数据很多,对于有需要的人而言,我觉得该方法这是不错的选择。
②.想下载全部转录因子结合位点数据过于困难,看似下载完了,其实是数据连接中断,它也不会报错,需要拉到txt最底部查看。因此,可能你已经有了感兴趣的转录因子,这个方法会比较合适,单独下载这个转录因子数据,来预测是否能和结合在目标基因启动子上。

下面开始介绍具体方法。
1.下载需要的物种基因组数据库,我们此处选择人。
首先打开Ensembl genome browser 106。
点击BioMart。

选择你所需要的基因组数据库。
选择完毕后,点击左侧的Attributes,按照下图左侧Attributes所示依次勾选需要下载的数据。(一定要按照这个顺序,因为下面会用到第几列,不是这个顺序的话列名会错!!!)

然后,点击Results按钮。
结果如下图所示。点击GO下载基因组数据库。这样就得到基因组数据了。

2.下载转录因子结合位点矩阵
点击New回到主页面
重新选择数据库,与上述一样,上面说的猜测,是因为选择的库(看名字,是有证据的)
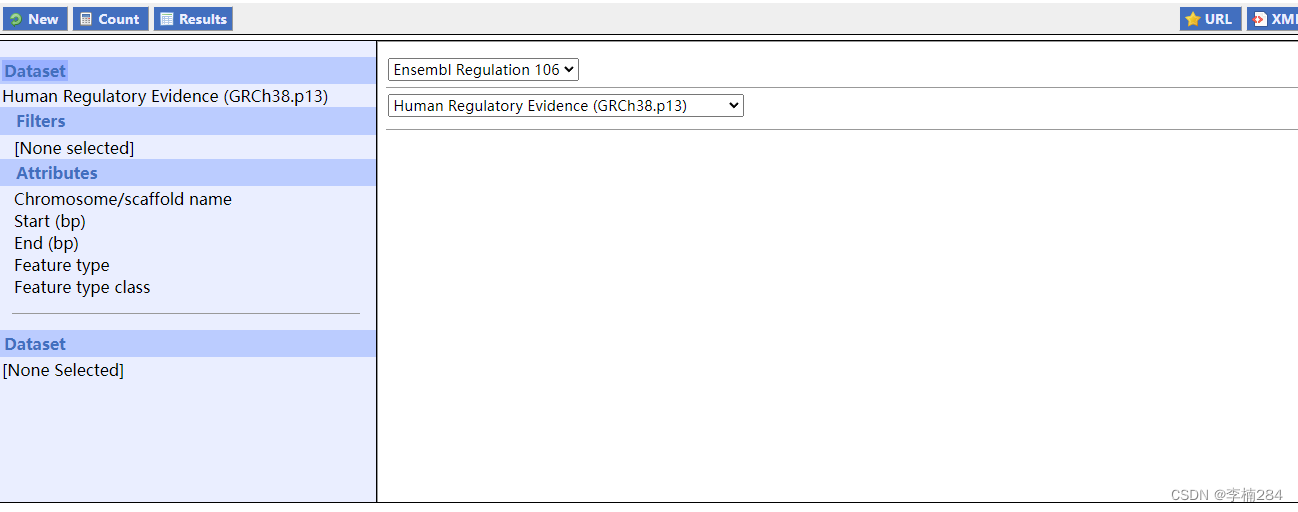
这边为了方便下载,我们通过Filters选择一下我们需要的数据。Attributes就按照图示自己按顺序选择一下。(如果后续有其他需求,这边过滤时, 自行选择即可。就是太难下载了!)

点击Results,然后点击GO下载。
3.下载完毕后,将它们转变为bed格式文件。其实,就是将文件名改一下。第一个命名为GRCh38.gene.bed。第二个命名为GRCh38.TFBS.bed。
然后,将两个文件放到/home/linan目录下。(当然,你要从桌面打开终端,你就把它放在桌面就行,这个自行选择。linan是我的用户名,改为自己用户名即可)
4.打开终端,查看下两个bed文件。代码分别是下面几行。发现文件没问题,我们继续下一步。
###查看开头文件
head GRCh38.gene.bed
head GRCh38.TFBS.bed
###查看末尾文件
tail GRCh38.gene.bed
tail GRCh38.TFBS.bed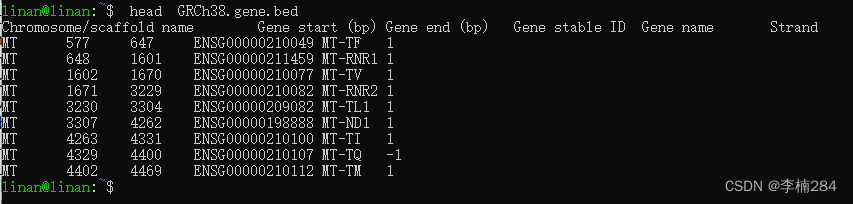

5.将基因组数据整理成我们想要的格式,主要是确定需要分析的启动子起始位置。
首先使用sed命令添加或删除需要的信息。
###删除第一行信息数据
sed -i '1d' GRCh38.gene.bed
###在Chromosome/scaffold name之前加上chr(个人感觉没啥意义)
###原来染色体正负链为-1或1,我们改为-和+
sed -i 's/-1$/-/' GRCh38.gene.bed
sed -i 's/1$/+/' GRCh38.gene.bed 文件格式就像这样即可。

然后,我们计算启动子-1000bp至+200bp的start和end位点。
###使用awk命令提取GRCh38启动子区域。命令含义为如果GRCh38.gene.bed的第6列“+”,那么以第2列Start (bp)为转录起始位点,并命名为TSS,我们-1000至+200bp只需要在TSS基础上-1000或者+200即可。如果是反向链(“-”),那么转录起始位点为第3列End (bp),将其命名为TSS,我们-1000至+200bp只需要在TSS基础上-200或者+1000(小白注意,因为3‘端是启动子区域,所以要End (bp)+1000。另外之所以小于0都计算为0,因为基因序列是从1开始计数的啊)。最终输出为GRCh38.gene.promoter.bed,其中第2,3列由我们计算结果替代。
awk 'BEGIN{OFS=FS="\t"}{if($6=="+") {TSS=$2; TSSUP=TSS-1000; TSSDOWN=TSS+200;} else {TSS=$3; TSSUP=TSS-200; TSSDOWN=TSS+1000;} if(TSSUP<0) TSSUP=0;print $1, TSSUP, TSSDOWN,$4,$5,$6;}' GRCh38.gene.bed > GRCh38.gene.promoter.bed使用head GRCh38.gene.promoter.bed查看计算结果。

6.处理下GRCh38.TFBS.bed文件。
这个简单,仅需使用sed命令去掉行名以及在Chromosome/scaffold name之前加上chr。
sed -i '1d' GRCh38.TFBS.bed
sed -i 's/^/chr/' GRCh38.TFBS.bed7.下载bedtools工具并利用它获得基因可能结合的转录因子。
sudo apt install bedtools下载即可。
利用bedtool合并两个数据,获得交集,并提取基因名、转录因子名至GRCh38.gene.promoter.TFBS.txt。
bedtools intersect -a GRCh38.gene.promoter.bed -b GRCh38.TFBS.bed -wa -wb | cut -f 5,10 | sort -u | tr 'a-z' 'A-Z' > GRCh38.gene.promoter.TFBS.txt8.匹配自己的基因集
将基因集放入/home/linan目录下,gene.txt。我们上一步基因名全是大写,以防万一,使用命令将基因集全部变为大写,并保存为list.text。cat gene.txt | tr "a-z" "A-Z" > list.txt
awk 'BEGIN{OFS=FS="\t"}ARGIND==1{save[$1]=1;}ARGIND==2{if(save[$1==1]) print $0}' list.txt GRCm39.gene.promoter.TFBS.txt > targetGene.TFBS.txt再次使用awk命令,匹配自己的基因集list.txt,到GRCh38.gene.promoter.TFBS.txt上,并将预测结果输出为targetGene.TFBS.txt。其实这边,由于txt的缘故,会匹配不到基因,因为多列数据,txt算一列,具体解决策略太菜了也没想到,只好用R语言的merge函数合并一下,效果一样。下图是我做的另外一个数据。

9.OK,到这里就结束啦,查看targetGene.TFBS.txt就行了。















 414
414











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









