






单细胞|Signac 进行 Motif 分析
引言
本教程将指导您如何在Signac平台上进行DNA序列的基序(Motif)分析。会介绍两种基序分析的方法:一种是在一组差异可访问的峰值中寻找出现频率较高的基序;另一种是在不同细胞群组间进行基序活性的差异分析。
library(Signac)
library(Seurat)
library(JASPAR2020)
library(TFBSTools)
library(BSgenome.Mmusculus.UCSC.mm10)
library(patchwork)
Data
本次演示[1]所使用的数据来源于成年小鼠的大脑。您可以在的教程中找到生成这些数据对象的代码以及原始数据的链接。首先,请先安装所需的软件包,并加载预先计算好的Seurat数据对象。
mouse_brain <- readRDS("adult_mouse_brain.rds")
mouse_brain
## An object of class Seurat
## 179011 features across 3512 samples within 2 assays
## Active assay: peaks (157203 features, 157203 variable features)
## 2 layers present: counts, data
## 1 other assay present: RNA
## 2 dimensional reductions calculated: lsi, umap
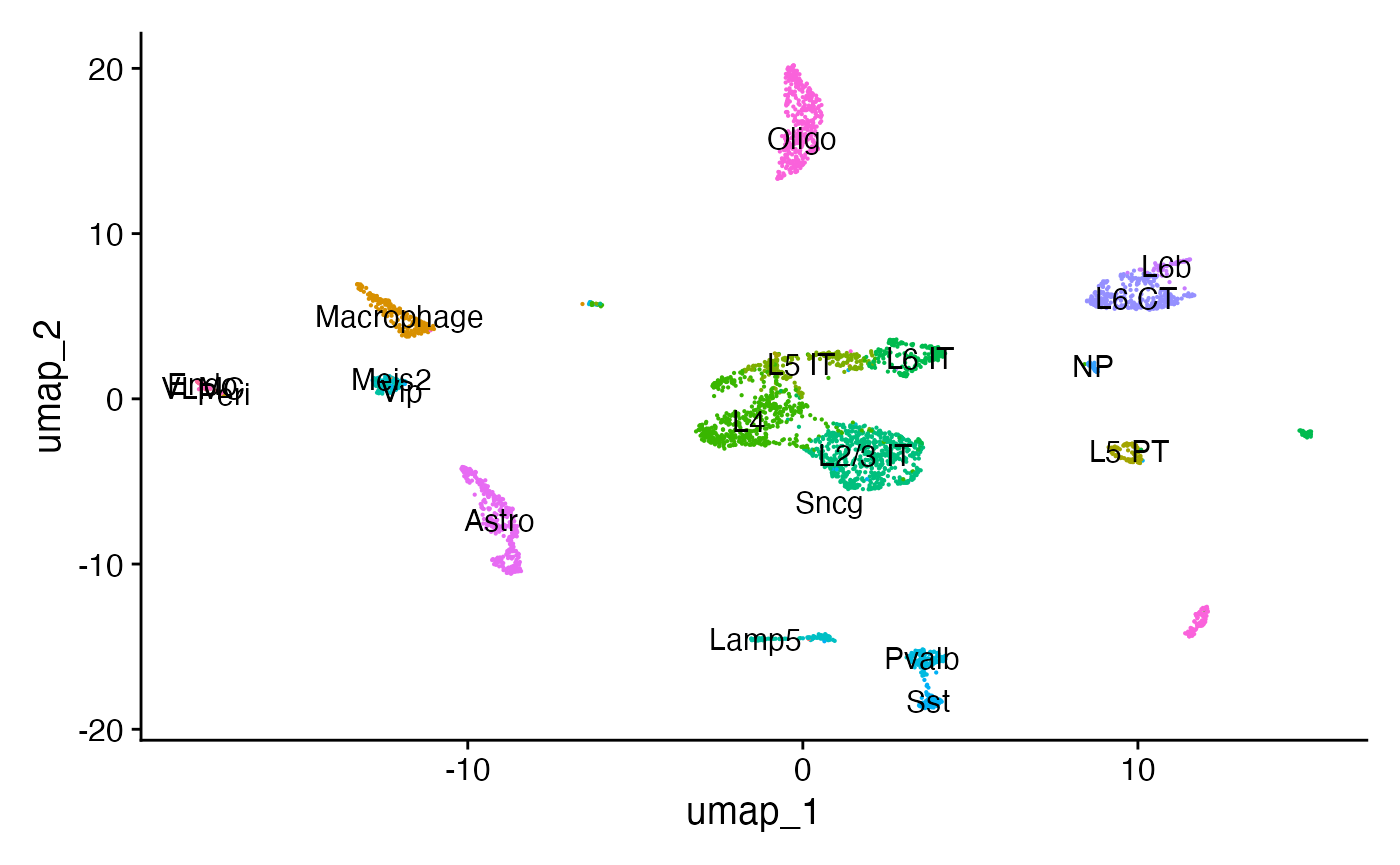
p1 <- DimPlot(mouse_brain, label = TRUE, pt.size = 0.1) + NoLegend()
p1
添加基序信息
要为基序分析添加所需的 DNA 序列基序信息,可以使用 AddMotifs() 函数来实现。
# Get a list of motif position frequency matrices from the JASPAR database
pfm <- getMatrixSet(
x = JASPAR2020,
opts = list(collection = "CORE", tax_group = 'vertebrates', all_versions = FALSE)
)
# add motif information
mouse_brain <- AddMotifs(
object = mouse_brain,
genome = BSgenome.Mmusculus.UCSC.mm10,
pfm = pfm
)
为了简化 Signac 平台上的基序分析工作,设计了一个名为 Motif 的类来存储所有必要的数据,这包括一系列位置权重矩阵(PWMs)或位置频率矩阵(PFMs),以及一个记录基序出现次数的矩阵。AddMotifs() 函数的作用是创建一个 Motif 对象,并将其整合到的小鼠大脑数据集中,同时还会包含每个峰的碱基组成等其他信息。任何 Seurat 检测都可以通过 SetAssayData() 函数添加 Motif 对象。
寻找过表达的基序
为了发现可能对细胞类型特异的调控序列有重要意义的基序,会寻找在不同细胞类型间差异可访问的峰集中出现频率过高的 DNA 基序。
在这个例子中,对比了 Pvalb 和 Sst 两种抑制性神经元的差异可访问峰。对于像单细胞染色质可及性测序(scATAC-seq)这样的稀疏数据,通常需要在 FindMarkers() 函数中调低 min.pct 的阈值,因为默认值(0.1)是针对单细胞 RNA 测序(scRNA-seq)数据设计的。
接下来,会进行超几何检验,以评估在特定频率下观察到某个基序是否仅仅是偶然现象,这需要与一组 GC 含量匹配的背景峰集进行比较。
da_peaks <- FindMarkers(
object = mouse_brain,
ident.1 = 'Pvalb',
ident.2 = 'Sst',
only.pos = TRUE,
test.use = 'LR',
min.pct = 0.05,
latent.vars = 'nCount_peaks'
)
# get top differentially accessible peaks
top.da.peak <- rownames(da_peaks[da_peaks$p_val < 0.005 & da_peaks$pct.1 > 0.2, ])
可选操作: 选定背景峰集 在寻找富集的 DNA 序列基序时,确保背景峰集的匹配性是至关重要的。通常,会选取一组与 GC 含量相匹配的峰作为背景峰集,但有时为了更精确地找到差异可访问的峰,将背景峰集限定为在特定细胞群组中可访问的峰可能会更有帮助。
AccessiblePeaks() 函数能够帮助识别出在某些细胞子集中开放的峰集。可以利用这个函数首先筛选出在 FindMarkers() 函数中用于比较的细胞群组里开放的峰集,作为潜在的背景峰集,然后通过 MatchRegionStats() 函数从这个更大的峰集中创建出一组与 GC 含量相匹配的峰集。
# find peaks open in Pvalb or Sst cells
open.peaks <- AccessiblePeaks(mouse_brain, idents = c("Pvalb", "Sst"))
# match the overall GC content in the peak set
meta.feature <- GetAssayData(mouse_brain, assay = "peaks", layer = "meta.features")
peaks.matched <- MatchRegionStats(
meta.feature = meta.feature[open.peaks, ],
query.feature = meta.feature[top.da.peak, ],
n = 50000
)
## Matching GC.percent distribution
Peaks.matched 然后可以通过在 FindMotifs() 中设置 background=peaks.matched 用作背景峰值。
# test enrichment
enriched.motifs <- FindMotifs(
object = mouse_brain,
features = top.da.peak
)
## Selecting background regions to match input sequence characteristics
## Matching GC.percent distribution
## Testing motif enrichment in 1131 regions

还可以绘制基序的位置权重矩阵,因此可以可视化不同的基序序列。
MotifPlot(
object = mouse_brain,
motifs = head(rownames(enriched.motifs))
)
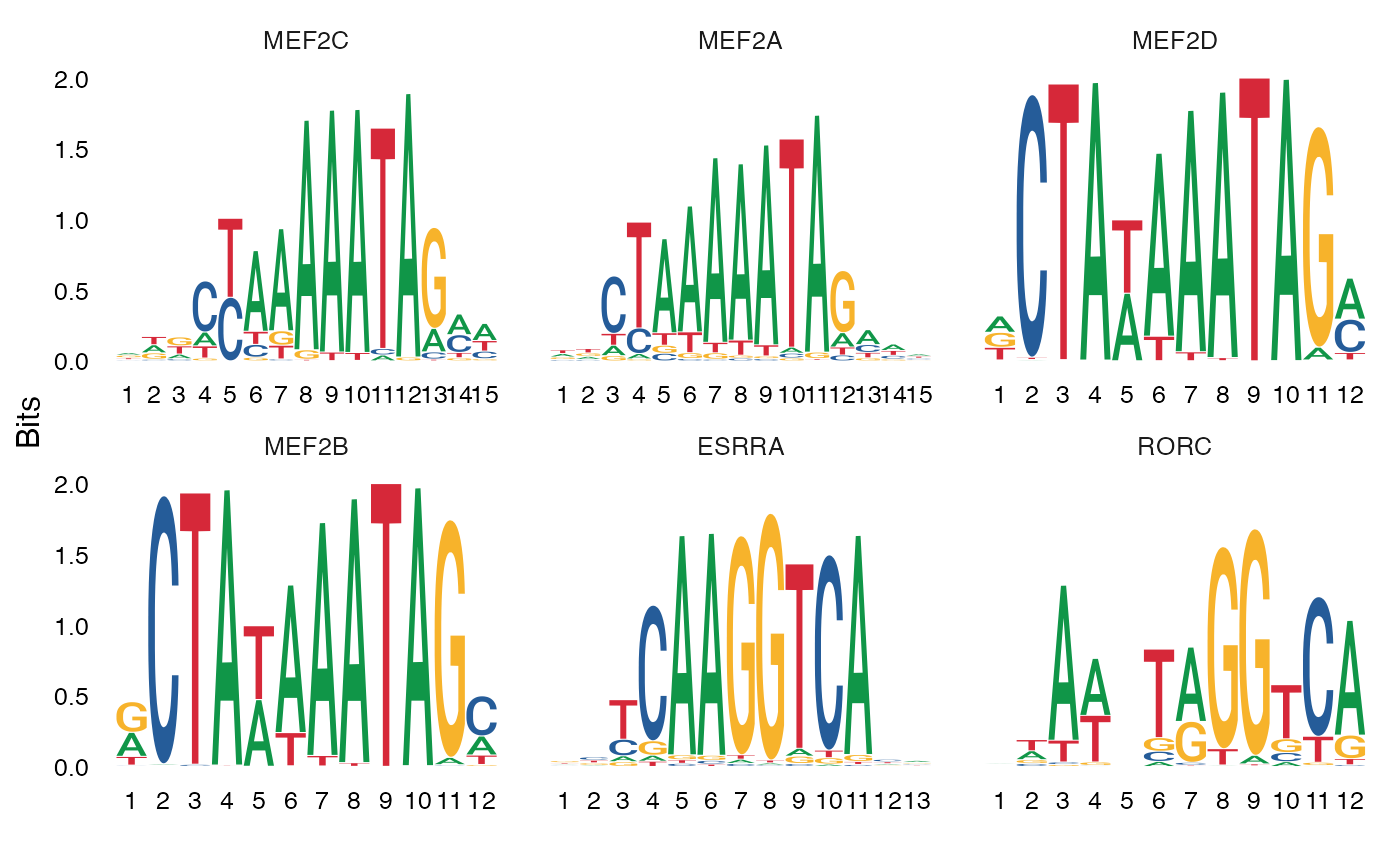
Mef 家族的基序,尤其是 Mef2c,在单细胞染色质可及性测序(scATAC-seq)数据中,与 Pvalb 特异性峰的关联性更强(相关研究请参考链接;链接),并且 Mef2c 对于 Pvalb 抑制性神经元的发育至关重要(相关研究请参考链接)。的研究结果与这些发现相吻合,并且在 FindMotifs() 函数的顶部结果中,发现 Mef 家族基序显著富集。
计算基序活性
还可以通过执行 chromVAR 分析来为每个细胞计算基序活性得分。这不仅使能够按细胞查看基序活性,还为提供了一种识别不同细胞类型中活性差异基序的新方法。
ChromVAR 能够识别与细胞间染色质可及性变化相关的基序。关于该方法的详细描述,请参阅 chromVAR 的相关论文。
mouse_brain <- RunChromVAR(
object = mouse_brain,
genome = BSgenome.Mmusculus.UCSC.mm10
)
DefaultAssay(mouse_brain) <- 'chromvar'
# look at the activity of Mef2c
p2 <- FeaturePlot(
object = mouse_brain,
features = "MA0497.1",
min.cutoff = 'q10',
max.cutoff = 'q90',
pt.size = 0.1
)
p1 + p2

还可以对不同细胞类型直接进行差异活性得分的测试。这种方法通常会得到与之前描述的在细胞类型间对比差异可访问峰的富集测试相似的结果。
在对 chromVAR 的 z 分数进行差异性分析时,可以在 FindMarkers() 函数中设置 mean.fxn 为 rowMeans,fc.name 为 "avg_diff",这样就能够计算出不同组之间 z 分数差异的平均值。
differential.activity <- FindMarkers(
object = mouse_brain,
ident.1 = 'Pvalb',
ident.2 = 'Sst',
only.pos = TRUE,
mean.fxn = rowMeans,
fc.name = "avg_diff"
)
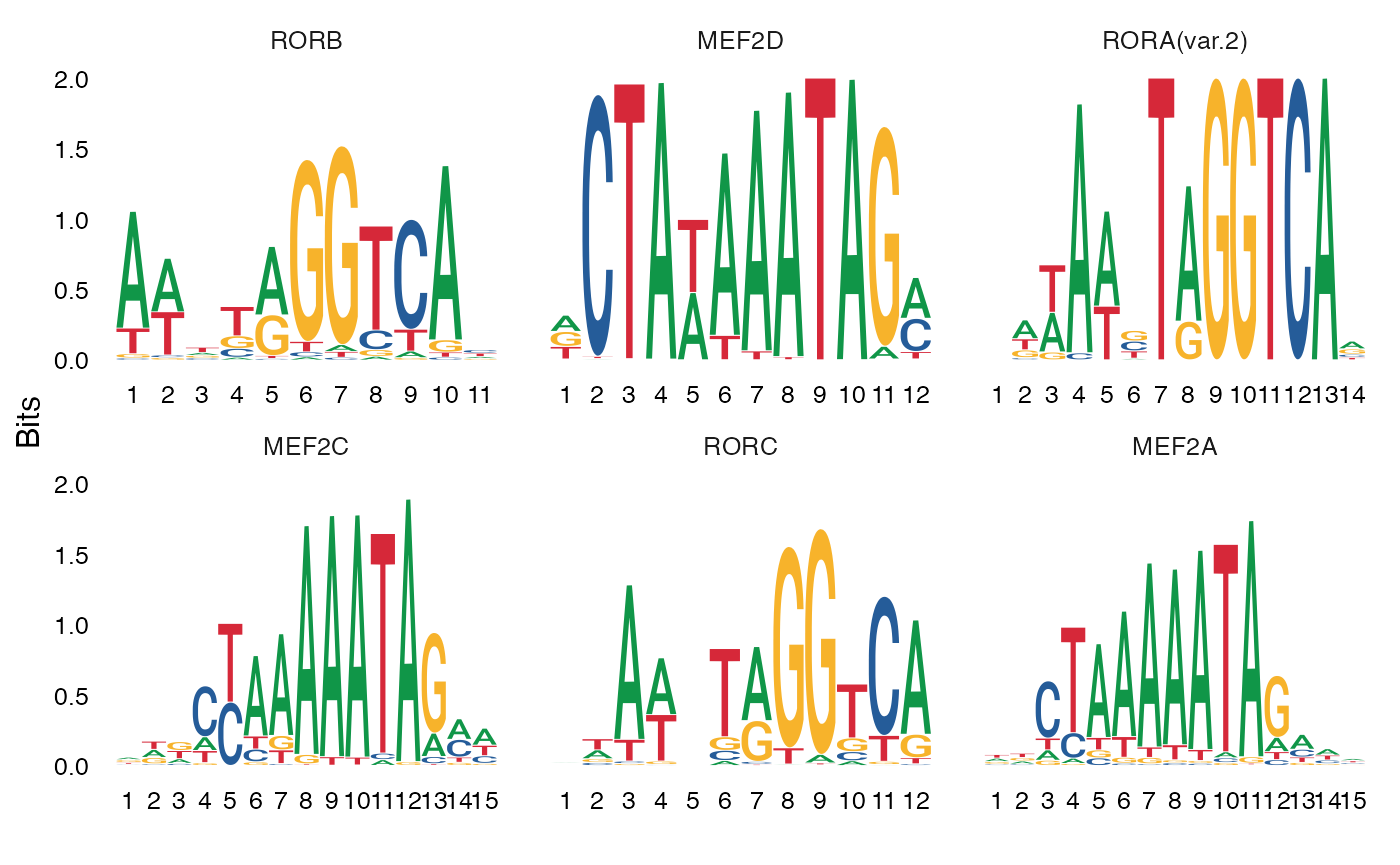
MotifPlot(
object = mouse_brain,
motifs = head(rownames(differential.activity)),
assay = 'peaks'
)
Source: https://stuartlab.org/signac/articles/motif_vignette
本文由 mdnice 多平台发布















 1243
1243

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









