






背景篇
MITE属于II类非自主转座因子,并且在真核生物中存在大量的拷贝。 MITE长度大约是500bp,并且扩增速度非常快。2013年一篇发表在NAR的文章"P-MITE: a database for plant miniature inverted-repeat transposable elements", 作者统计了各个物种中MITE的数目
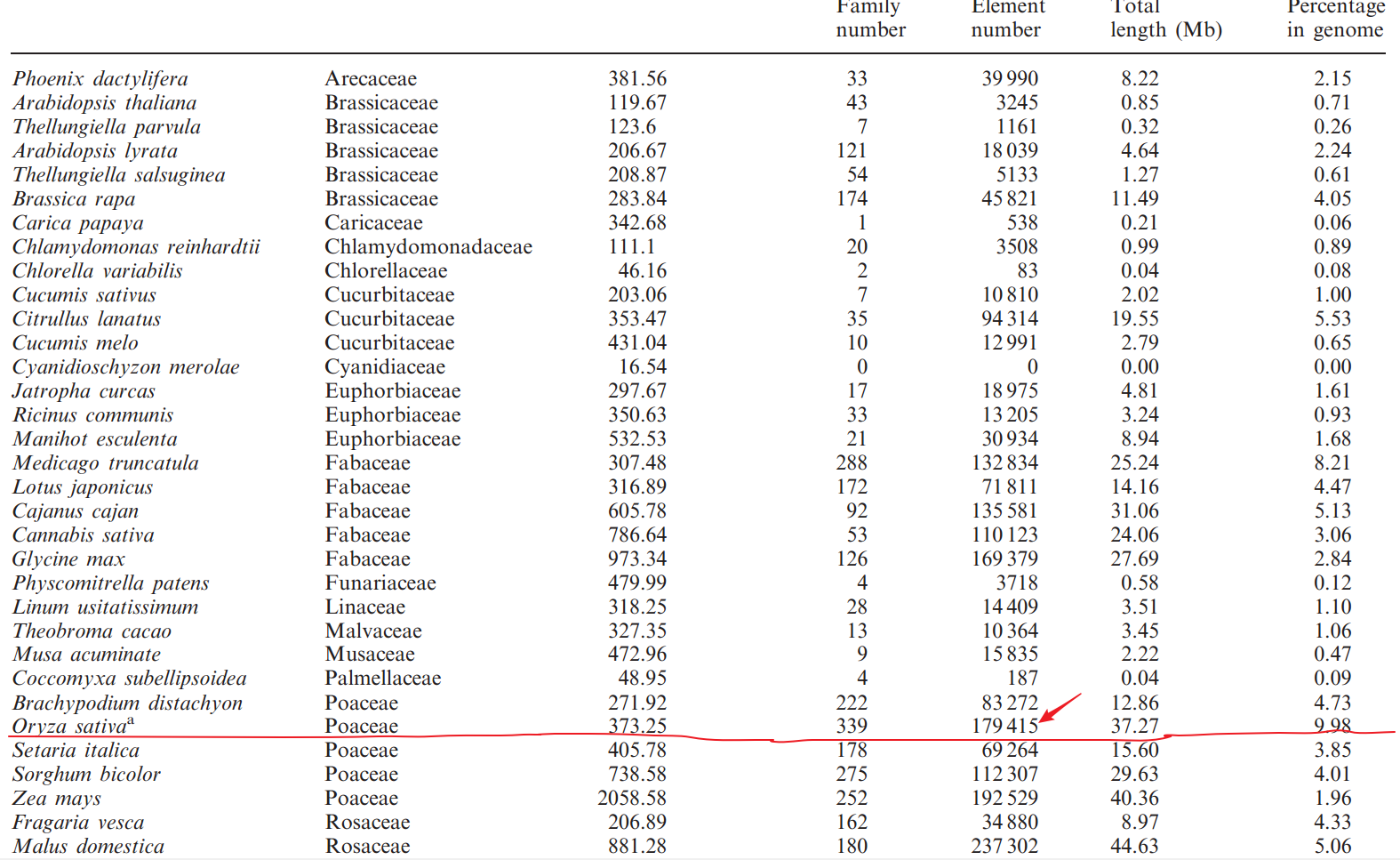
目前主要有两大家族,Stowaway和Tourist,分别来自于Tc1/Mariner和PIF/Harbinger超家族。一个典型MITE结构如下:
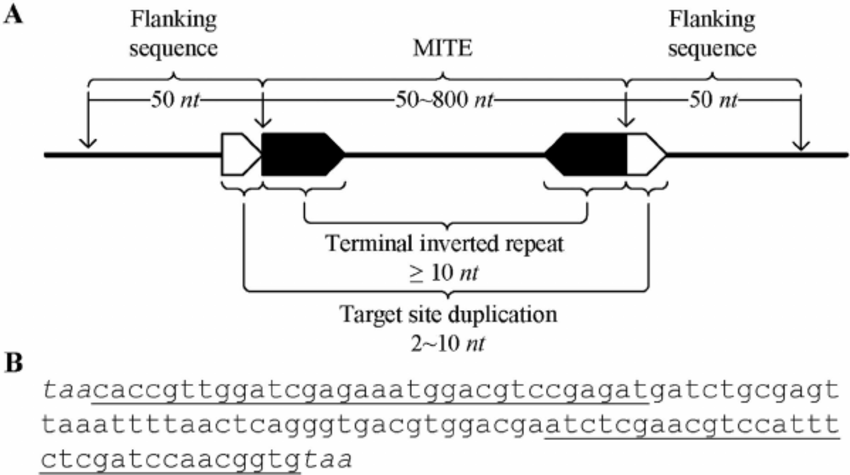
MITE-Hunter从基因组上搜索II类转座因子,如MITE(miniature inverted repeat transposable elements),以及长度低于2kb的非自主转座因子。MITE-Hunter的分析流程如下图所示,主要分为五步:
- 根据MITE的结构特征搜索候选的TE
- 通过配对序列联配(Pairwise Sequence Alignment, PSA) 过滤假阳性
- 得到模板序列
- 基于多序列比对(Multiple Sequence Alignment, MSA)进一步过滤假阳性,构建一致性序列,并预测TSD
- 将一致性序列进行分组,归类到不同家族。
注:
- TIR: terminal inverted repeats 末端反向重复
- TSD: target site duplication 靶点重复
安装篇
安装MITE-Hunter之前,先要安装其他三个软件:
- NCBI BLAST: 下载链接为https://ftp.ncbi.nlm.nih.gov/blast/executables/legacy.NOTSUPPORTED/2.2.9/,注意选择平台
- Muscle: 下载地址为http://www.drive5.com/muscle/downloads.htm
- mDust: 下载地址为https://github.com/lh3/mdust
从http://target.iplantcollaborative.org/mite_hunter.html下载,进入解压后的文件夹中,运行如下安装命令
perl MITE_Hunter_Installer.pl \
-d /opt/biosoft/MITE_Hunter/ \ #MITE_hunter解压缩后的文件夹路径
-f /opt/biosoft/blast-2.29/formatdb \ # formatdb的路径
-b /opt/biosoft/blast-2.29/blastall \ #blastall的路径
-m /opt/biosoft/mdust/mdust \ # mdust的路径
-M /opt/biosoft/muscle/muscle #muscle的路径
使用篇
下面的操作中,假设你下载了拟南芥的基因组,并且命名为TAIR10.fa
MITE-Hunter只要求单个输入文件,但是有很多参数需要调整。
perl MITE_Hunter_manager.pl \
-i TAIR10.fa \
-g thaliana \
-n 5 \
-S 12345678 \
-P 1 &
参数说明:
核心的三个参数:
- -i 输入的基因组序列
- -P:使用多少比例的序列去搜索TE,对于700Mb以下的基因组用1. 参数可以设置为1/(实际基因组大小/700),例如人类基因组是3G, 那么就可以是0.25.
- -g: 输出文件名的前缀
其他参数, 除了改改线程数以外,基本上都是无脑用作者的默认参数。
- -w: 最大能发现的TE长度,默认是2000
- -c: CPU数, 默认是5
- -n: 最多有多少组,默认5.
- -d: 这个参数过滤低复杂度序列,例如"AAAAA...", "TATATATATA...", "GGGG..", 默认是0.2,也就是预测TE序列要是超过20%。后续还有一个 -p 参数和该参数一样,不知道作者是怎么想的。
- -f: MITE两翼的序列长度(默认60), 用于判断TE是否为真。
- -t: 最短TIR(terminal inverted repeat)的长度, 默认10,用于寻找候选的TE
- -M: TSD(the longest target site duplication)的长度,默认10,用于寻找候选的TE
- -l: TIR区域所允许的最大错配碱基数, 默认1.
- -L: 默认是 90, 表示两个TE至少有 90 bp 相似的序列,才会被归为一组
- -I: 默认是 80, 表示两个TE要是 80% 以上的相似度就会被归为一组。
- -m: TE最多少要有多少拷贝数,默认是3.
- -T: 作者不推荐你修改。参数默认是"TA_"表示候选TE必须包含2bp即"TA"的TSD。
- -C: 默认0,表示MITE_Hunter会使用找到的TE的全部序列进行搜索,寻找是否有其他拷贝,如果设置为1,则表示只用前后200bp去搜索。
- -A: 默认是90,表示如果TE中有超过90bp的低复杂度序列,则过滤。
- -S: MITE-Hunter一共有8步,你可以用"12"先只运行前2步,然后用"345678"运行后续的几步。
- -F : 0 或1, 默认MITE_hunter会自动处理
- -s: 没有具体说明作用
输出文件
MITE-Hunter的输出文件包括分组后的一致性TE序列及其对应多重联配文件。其中以".aln.elite"结尾的文件便是多重序列联配结果文件(MSA)。文件名中有"Step8_"的文件则包含TE一致性序列,每个文件都是一个TE家族,除了"Step_8.singlet"和"Step_8.paired". 前者里的TE在基因组上没有相似的同源序列,后者是潜在的复合TE序列。
你还可以手动检查输出结果,过滤一些假阳性。例如通过检查MSA文件,根据能否确定TIR和TSD的位置判断预测的TE是否真实存在

以及将预测的TE在http://target.iplantcollaborative.org/进行检索,判断一个TE是否是有多个其他TE组成。
最后合格的序列,或者直接将输出文件,Step8_*.fa” 和 “Step8_singlet.fa”候选的MITE序列,你可以将其命名为MITE.lib,用作后续的RepeatMasker输入.
推荐阅读
- MITE-Hunter: a program for discovering miniature inverted-repeat transposable elements from genomic sequences
- MITE-Hunter_manual
- P-MITE: a database for plant miniature inverted-repeat transposable elements
其他的一些MITE软件: MITE Digger, RSPB

















 1325
1325

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









