






1.鉴定
将转座子鉴别和注释的方法分为3大类:
①从头算法;
②基于同源性的方法 ;
③联合算法。
其中,从头算法又包括基于基因组序列比对的方法、K—mer方法和基于结构特征的方法 ;
基于同源性的方法包括基于同源序列比对的方法和基于隐马尔柯夫模型(HMM)的方法。
不同的算法实现的目标有所不同,如从头算法主要是为了发现新的转座子 ,因此常用来在新测序的基因组中鉴别新的转座子 ,而多数基于同源性算法的软件主要被用来注释基因组中的转座子。
从头( De novo ) 算法
-
从头算法鉴别转座子的原理是基于转座子的重复特征,该算法可在不借助任何转座子数据库的情况下查找到几乎所有类型的转座子。优点:算法主要用于发现新的、未注释的转座子家族, 对于高频出现的转座子鉴别尤其有效。缺点:计算量大。另外, 由于从头算法是基于用一个转座子的拷贝数来定义重复家族, 这样低拷贝数的转座子可能被忽略掉。根据算法原理的不同, 从头算法又包括基于基因组序列比对的方法、K-mer 方法和基于结构特征的方法。
-
基于基因组序列比对的方法。基于序列相似性,该方法利用 BLAST 等软件将基因组与基因组进行比对, 然后将双序列比对的结果转换成多序列比对, 最后用聚类方法将相关序列聚成家族,从而得到重复序列( 包括转座子) 家族。代表:RECON
-
K-mer 方法。 该类从头算法检索重复出现的定长 Kmer 种子( 序列短串) , 然后再将它扩展为更长的序列。代表:RepeatScout。软件首先在未知的基因组序列中计算出所有定长 K-mer 种子出现的频率, 再选择出最高频率的 K-mer 种子及其周围区域的序列, 一次一个碱基向两边扩展, 每次生成一条具有代表性 K-mer 重复家族的共有序列。然后调整已出现过的 K-mer 频率数, 再选择出包括调整过的最高频率 K-mer 及其周围区域序列, 扩展并产生共有序列,直到最高频率到达所设定的最小阈值结束, 这样就得到了这一基因组的转座子家族。RepeatScout与 RECON 相比, 所得到结果更加准确,而且敏感度和运行速度都有很大的提高。其他使用 K-mer 方法的软件还有RepeatFinder等。
-
基于结构特征的算法。 转座子中 LTR 逆转录转座子、SINE、微型反向重复转座元件( MITE) 、Helitron 等都具有较明显的特征, 基于结构特征的算法可根据这些特征对这些转座子进行鉴别。代表:LTR_STRUC,LTR_FINDER,LTRharvest ,LTRdigest。LTR_finder 和 LTRharvest 是目前为止鉴定 LTR 最敏感的程序,但假阳性依然很高。
基于同源性的算法
- 基于同源性的算法是将一条未知序列与已知的转座子序列或序列特征模型进行比较,从而鉴别转座子的一类方法。根据同源序列比较方法的不同, 基于同源性的方法又可分为基于同源序列比对的算法和基于 HMM 的算法 2 类。
- 基于同源序列比对的算法。该类算法与从头算法中的基于基因组序列比对的方法都是使用 BLAST 等工具来发现序列相似性, 但与后者不同的是, 基于同源序列比对的方法是将未知序列与数据库中的转座子序列进行比较来鉴别转座子。转座子数据库可使用公共数据库 Repbase, 但现在自己物种的研究,基本都是通过当前的全基因组序列,训练重复序列集构建本地repeat library,再通过RepeatMasker注释重复序列。其中,与RepeatMasker配套的RepeatModeler,可以实现。
代表:RepeatMasker。RepeatMasker 利用 BLAST 工具在转座子数据库(Repbase或者自己构建的repeat library)中比对查找已知的重复因子家族, 是目前基因组转座子注释最常用的软件。 - 基于 HMM 的算法。中文论述见 https://kns.cnki.net/kcms/detail/detail.aspx?dbcode=CJFD&dbname=CJFD2013&filename=AHNY201308003&uniplatform=NZKPT&v=%25mmd2Fe0g9nSKUuUK1GjQz6eD4Op0avSl%25mmd2BYGyQTsPxiKcx27YQ9yw%25mmd2FVAPSWnHERxDx8gt
数据库
- RepBase Update (https://www.girinst.org/repbase/update/) 是检索真核生物基因组中各类移动元件/转座元件共有序列集的最常用的数据库之一。数据库旨在给出每一类TE家族的共有序列和代表型元件类型。它将转座元件分为三类:DNA转座子,LTR逆转录转座子(Long Terminal Repeat Retrotransposons)和non-LTR逆转录转座子。
- Dfam是一个较RepBase更“年轻”的真核生物TE-centric数据库,这个数据库更正式地定义了转座元件,并且将共有序列一样的转座元件形成一个“集合”,利用隐马尔可夫模型(hidden Markov models)来进行多序列比对(multiple sequence alignments)。
- TREP,一个可用于研究植物和真菌中转座元件的数据库。这个存储库有两个子库,一个包含不同TE元件子类的共有序列(nrTREP),另一个包含单个插入的TE元件的完整序列(total_TREP);同时这个存储库中还有一个数据库是对那些插入序列进行蛋白预测的(PTREP)。
- P-MITE是一个植物特有的数据库,其中MITErepdb主要包含共有序列信息,而MITEdb主要用于注释41种植物基因组中的各个微型倒置重复转座元件(miniature inverted repeat TE, MITE)。
- RiTE是特别用于水稻及其相关物种基因组中重复元件研究的数据库,这个数据库中包含水稻基因组中全部转座元件的序列,共有序列以及单个转座子在参考基因组中的插入信息。
- MASiVEdb数据库包含和RiTE差不多的信息,只是MASiVEdb包含更多的植物物种的转座子注释信息。
- 还有一些数据库是对那些插入到基因组上各基因的编码区的转座元件做汇总,包括TranspoGene, HESAS, 以及LINE FUSION GENES。
- RepeatExplorer database (REXdb):https://link.springer.com/article/10.1186/s13100-018-0144-1 REXdb根据来自于80个植物物种保守的多蛋白结构域,将Copia和Gypsy反转录转座子分别分为16个和14个谱系。
RepeatModeler,LTR_retriever
-
RepeatModeler 利用全基因组序列从头预测(de novo),训练重复序列集构建本地 repeat library。
RepeatModeler1.0 核心组件是 RECON(de novo,基于基因组序列比对)和 RepatScout (de novo,基于 K-mer)。RepeatModeler2.0更新后加入了 LtrHarvest(de novo,基于结构),LTR_retriever(主要有LTR_FINDER,LTRharvest,都是 de novo 基于结构) 等,可以识别LTR的结构。 -
公司的重复序列注释流程:使用RepeatModeler从头鉴定(都是 de novo,1.0基于基因组序列比对,基于 K-mer;2.0 基于基因组序列比对,基于 K-mer,基于结构)重复区域家族,生成repeat library。然后再用RepeatMasker(基于同源性)鉴别基因组上的重复区域。
-
用 LTRharvest 和 LTRdigest 进行 LTR 基于结构 的从头预测( de novo,基于结构 )
-
2017 年密歇根州立大学园艺系的 Shujun Ou 团队开发 LTR_retriever 平台用于 LTR 的鉴定,文章发表在 Plant Physiology 上。这是一款整合软件,以一或多个 LTR 预测软件鉴定 LTR 的结果作为输入文件,通过不同模块对 LTR 进行过滤和修正来对预测软件的预测结果进行整合和调整,得到非冗余精准且完整的物种特异 LTR 库,再使用 RepeatMasker 进行预测。
LTR_retriever不是一个独立的工具,他的主要作用就是整合 LTRharvest, LTR_FINDER, MGEScan 3.0.0, LTR_STRUC, 和 LtrDetector的结果,过滤其中的假阳性LTR-RT,得到高质量的LTR-RT库。尽管LTR_retriever支持多个LTR工具的输入,但其实上LTRharverst和LTR_FINDER的结果就已经很不错了。目前推荐的是LTR_Finder(de novo,基于结构)和LTR_harvest(de novo,基于结构)组合鉴定,之后使用LTR_retreiver整合两者的结果。
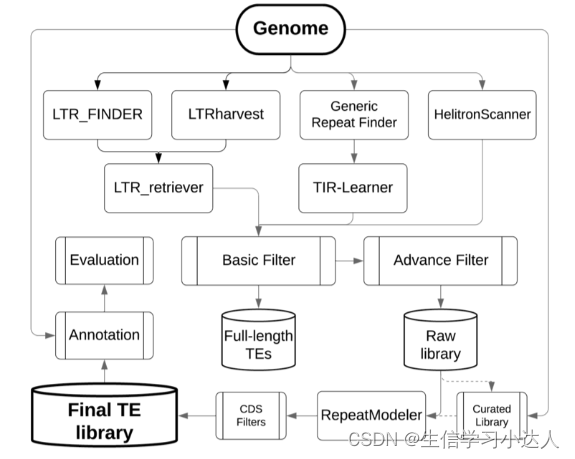
EDTA, 全称是 Extensive de-novo TE Annotator, 一个综合性的流程工具,它整合了目前LTR预测工具结果(LTR_retriever,基于结构),TIR预测工具结果,MITE预测工具结果,Helitrons预测工具结果,从而构建出一高可信,非冗余的TE数据库,再通过 RepeatMasker(基于同源性) 注释重复序列。
EDTA的下载安装及使用方法参考 EDTA-github
2. 重复序列的校正和分类
常用的软件TEclass(Institute of Bioinformatics WWU Münster)、REPCLASS(http://wweb.uta.edu/faculty/cedric/repclass.htm)等
3. 基因组注释(重复序列的屏蔽)
重复序列库经过校正和分类以后, 就可以用于全基因组重复序列注释。这个过程被称为重复序列屏蔽, 鉴定为重复序列的核苷酸区域常用N或者X替 代。常用的软件有RepeatMakser(RepeatMasker Home Page)和CENSOR(Submit sequence to CENSOR - GIRI)等, 详细的使用说明请参考。此过程在很大程度上依赖于重复序列库的质量 , 例如一致序列的准确性及分类的准确性等。
参考:
TE的鉴定 - 简书

















 2686
2686

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









