






使用ALLHiC基于HiC数据辅助基因组组装
基因组组装大致可以分为三步(1)根据序列之间的重叠情况构建出contig,(2)基于二代的mate pair文库或光学图谱将contig搭建成scaffold,(3)对scaffold进行排序和调整方向得到最终的准染色体级别的基因组。
目前的三代测序组装能够搞定第一步和第二步。而在将contig/scaffold提升至准染色体水平上,有4种方案可选。一种是基于遗传图谱,一种是利用BioNano DLS光学图谱,一种是利用近缘物种的染色体同源性,还有一种就是HiC。其中HiC技术是三者中较为简单的一个,不需要高质量的DNA文库,也不需要一个很大的群体,结果也比较准确可信。
HiC的文库构建示意图如下,我们所需要的就是最终双端测序的两端序列之间的距离关系。

目前利用HiC数据进行组装软件有 LACHESIS, HiRise, SALSA1, 3D-DNA等,这些软件在动物基因组上和简单植物基因组上表现都不错,但是不太适合直接用于多倍体物种和高杂合物种的组装上。主要原因就是等位基因序列的相似性,使得不同套染色体之间的contig出现了假信号,最终错误地将不同套染色体的contig连在了一起。最近在Nature Plants发表的ALLHiC流程就是用来解决多倍体物种和高杂合度基因组的HiC组装难题。
ALLHiC流程一览
ALLHiC一共分为五步(见下图,Zhang et al., 2019),pruning, partition, rescue, optimization, building,要求的输入文件为HiC数据比对后的BAM和一个Allele.ctg.table。
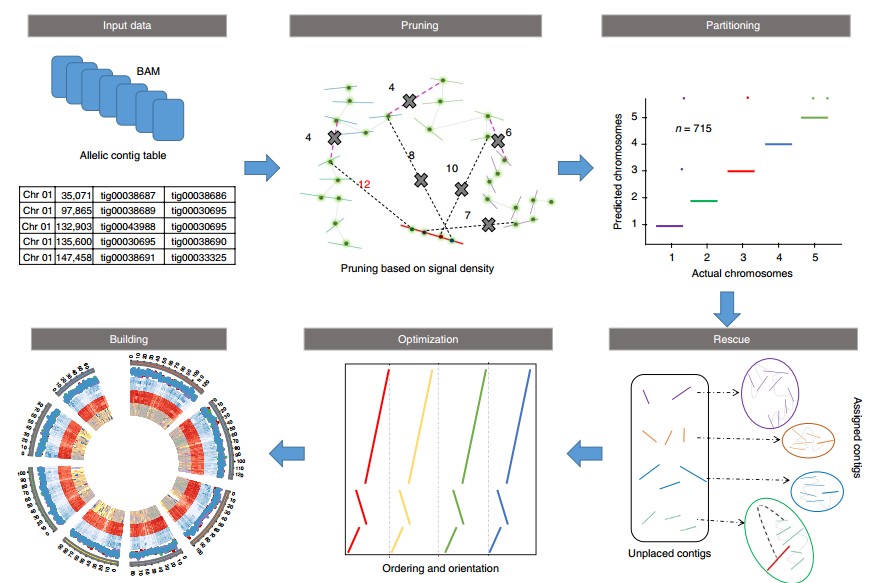
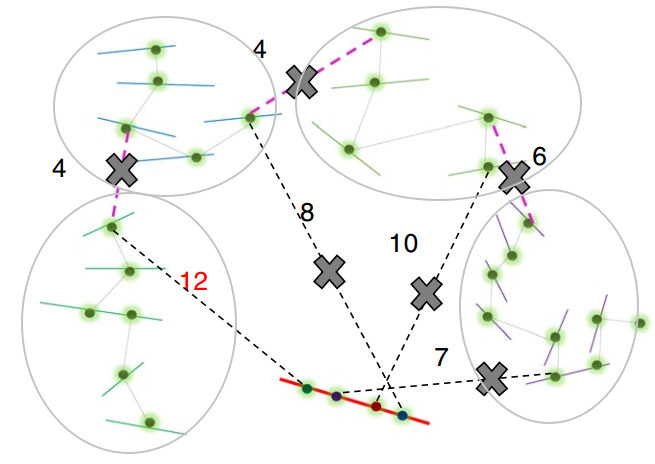
其中pruning步骤是ALLHiC区别于其他软件的关键一步。因此我专门将其挑选出来进行介绍,红色实线是潜在的坍缩区域(组装时因为序列高度相似而没有拆分),而其他颜色实线则是不同的单倍型(我用浅灰色椭圆进行区分)。粉红色虚线指的是等位基因间的HiC信号,而黑色虚线则是坍缩区域和未坍缩区域的HiC信号。
ALLHiC在这一步会根据提供的Allele.ctg.table过滤BAM文件中等位基因间的HiC信号,同时筛选出坍缩区域和未坍缩区域的HiC信号。这些信号会用于Rescue步骤,将未锚定contig分配到已分组的contigs群。
软件安装
ALLHiC的安装非常简单,按照习惯,我将软件安装在~/opt/biosoft下
mkdir -p ~/opt/biosoft && cd ~/opt/biosoft
git clone https://github.com/tangerzhang/ALLHiC
cd ALLHiC
mv allhic.v0.9.8 bin/allhic
chmod +x bin/*
chmod +x scripts/*
# 添加到环境变量
PATH=$HOME/opt/biosoft/ALLHiC/scripts/:$HOME/opt/biosoft/ALLHiC/bin/:$PATH
export PATH
此外ALLHiC还依赖于samtools(v1.9), bedtools 和 Python 3环境的matplotlib(v2.0+),这些可以通过conda一步搞定。
conda create -y -n allhic python=3.7 samtools bedtools matplotlib
之后检查下是否成功安装
$ conda activate allhic
$ allhic -v
$ ALLHiC_prune
你可能会遇到如下的报错
ALLHiC_prune: /lib64/libstdc++.so.6: version `GLIBCXX_3.4.21' not found (required by ALLHiC_prune)
这是GLIBC过低导致,但是不要尝试动手去升级GLIBC(你承担不起后果的),conda提供了一个比较新的动态库,因此可以通过如下方法来解决问题
export LD_LIBRARY_PATH=$LD_LIBRARY_PATH:~/opt/miniconda3/lib
ALLHiC分析实战
感谢张兴坦老师提供的测试数据,我将其中的BAM转成了原始FastQ格式,以便从头讲解。数据在百度网盘,可以在我的博客 对应的位置找到链接。
输入文件需要有4个,contig序列,等位contig信息和两个双端测序数据
$ ls -1
Allele.ctg.table
draft.asm.fasta
reads_R1.fastq.gz
reads_R2.fastq.gz
第一步: 建立索引
samtools faidx draft.asm.fasta
bwa index -a bwtsw draft.asm.fasta
第二步: 序列回贴。这一步的限速步骤是bwa sampe,因为它没有多线程参数。如果数据量很大,可以先将原始的fastq数据进行拆分,分别比对后分别执行bwa sampe,最后合并成单个文件。
bwa aln -t 24 draft.asm.fasta reads_R1.fastq.gz > reads_R1.sai
bwa aln -t 24 draft.asm.fasta reads_R2.fastq.gz > reads_R2.sai
bwa sampe draft.asm.fasta reads_R1.sai reads_R2.sai reads_R1.fastq.gz reads_R2.fastq.gz > sample.bwa_aln.sam
第三步: SAM预处理,移除冗余和低质量信号,提高处理效率
PreprocessSAMs.pl sample.bwa_aln.sam draft.asm.fasta MBOI
# 如果已有BAM文件
# PreprocessSAMs.pl sample.bwa_aln.bam draft.asm.fasta MBOI
filterBAM_forHiC.pl sample.bwa_aln.REduced.paired_only.bam sample.clean.sam
samtools view -bt draft.asm.fasta.fai sample.clean.sam > sample.clean.bam
其中filterBAM_forHiC.pl的过滤标准是比对质量高于30(MQ), 只保留唯一比对(XT:A:U), 编辑距离(NM)低于5, 错误匹配低于(XM)4, 不能有超过2个的gap(XO,XG)
第四步(可选): 对于多倍体或者是高杂合的基因组,因为等位基因的序列相似性高,那么很有可能会在不同套基因组间出现假信号,因此需要构建Allele.ctg.table, 用于过滤这种假信号。
ALLHiC_prune -i Allele.ctg.table -b sample.clean.bam -r draft.asm.fasta
这一步生成prunning.bam用于后续分析
第五步: 这一步是根据HiC信号将不同的contig进行分组,分组数目由-k控制。如果跳过了第四步,那么可以直接用第三步的结果sample.clean.bam
ALLHiC_partition -b prunning.bam -r draft.asm.fasta -e AAGCTT -k 16
这一步会生成一系列以prunning开头的文件
- 分组信息: prunning.clusters.txt
- 每个分组对应的contig: prunning.counts_AAGCTT.XXgYY.txt:
- 每个contig长度和count数: prunning.counts_AAGCTT.txt
第六步: 将未锚定的contig分配已有的分组中。
ALLHiC_rescue -b sample.clean.bam -r draft.asm.fasta \
-c prunning.clusters.txt \
-i prunning.counts_AAGCTT.txt
这一步根据之前prunning.counts_AAGCTT.XXgYY.txt对应的groupYY.txt
第七步: 优化每一组中的contig的排序和方向
# 生成.clm文件
allhic extract sample.clean.bam draft.asm.fasta --RE AAGCTT
# 优化
for i in group*.txt; do
allhic optimize $i sample.clean.clm
done
这一步会基于groupYY.txt生成对应的groupYY.tour
第八步: 将tour格式转成fasta格式,并生成对应的agp。
ALLHiC_build draft.asm.fasta
这一步生成两个文件,groups.asm.fasta和groups.agp。其中groups.asm.fasta就是我们需要的结果。
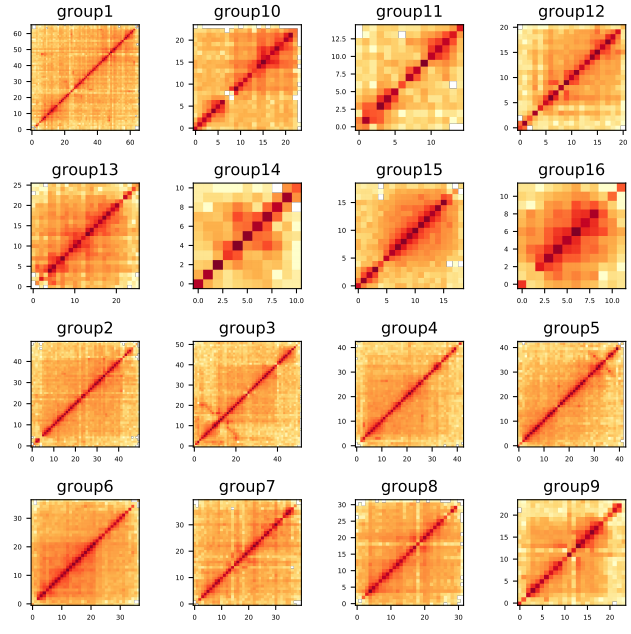
第九步: 构建染色质交互矩阵,根据热图评估结果
samtools faidx groups.asm.fasta
cut -f 1,2 groups.asm.fasta.fai > chrn.list
ALLHiC_plot sample.clean.bam groups.agp chrn.list 500k pdf
使用ALLHiC的几个注意事项:
- ALLHiC依赖于初始的contig,如果嵌合序列和坍缩序列比例过高,那么ALLHiC结果也会不准确。根据文章,ALLHiC能够处理
~10%的嵌合比例,~20%的坍缩比例。因此最好是用类似于Canu这种能够区分单倍型的组装软件。 - 单倍型之间序列相似度不能太高,否则会出现大量非唯一比对,降低可用的HiC信号
- 构建Allele.ctg.table需要一个比较近缘的高质量基因组
- 不要用过短的contig,因为短的contig信号少,很容易放到错误的区域
- K值的设置要根据实际的基因组数目设置,如果你发现输出结果中某些group过大,可以适当增大k值
参考资料
- https://github.com/tanghaibao/allhic
- https://github.com/tangerzhang/ALLHiC/wiki
- Zhang, X., Zhang, S., Zhao, Q., Ming, R., and Tang, H. (2019). Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data. Nat. Plants 5, 833–845.
- Zhang, J., Zhang, X., Tang, H., Zhang, Q., Hua, X., Ma, X., Zhu, F., Jones, T., Zhu, X., Bowers, J., et al. (2018). Allele-defined genome of the autopolyploid sugarcane Saccharum spontaneum L. Nature Genetics 50, 1565.
版权声明:本博客所有文章除特别声明外,均采用 知识共享署名-非商业性使用-禁止演绎 4.0 国际许可协议 (CC BY-NC-ND 4.0) 进行许可。


















 1728
1728

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









