







定义
之前的文章中有介绍过,HiC常用的几款软件的原理内容。可以点击链接访问了解一下
在这里不做赘述。
软件安装
新版本
建议使用目前最新的3.0.0版本(需要root权限)
安装方法如下:
# 创建conda环境
conda create -y -n hic-pro python=3.7 pysam bx-python numpy scipy samtools bowtie2 iced
# 下载HiC-Pro最新版本
wget https://hub.fastgit.org/nservant/HiC-Pro/archive/refs/tags/v3.0.0.tar.gz
tar zxf v3.0.0.tar.gz
cd HiC-Pro-3.0.0/
# 需要root权限
sudo make install
旧版本
如果新版本不适应可以使用旧版本
conda install -c davebx hicpro
其他安装方法参考官方
软件操作流程
数据准备
-
将Hi-C数据放入对应以样品名命名的目录下。
(注意文件夹名、_R1.fastq.gz._R2.fastq.gz) -
基因组组装结果文件
ln -s PATH=your_assembly.fasta genome.fa
必备文件1-基因组bowtie2索引
bowtie2-build genome.fa genome
酶切片段文件
/home/lixingze/software/HiC-Pro-3.0.0/bin/utils/digest_genome.py genome.fa -r dpnii -o genome_dpnii.bed
基因组中序列大小文件
samtools faidx genome.fa
awk '{print $1"\t" $2}' genome.fa.fai >genome.sizes
运行hic-pro
HiC-Pro -c config-hicpro.txt -o analysis -i data
config-hicpro.txt 配置
其中没有提到的建议使用默认数据操作
## SYSTEM AND SCHEDULER - Start Editing Here !!
N_CPU = 50 #CPU线程数
LOGFILE = hicpro.log #log文件名
JOB_NAME = sample #任务名
JOB_MEM = 100gb #占用内存
JOB_WALLTIME =
JOB_QUEUE =
JOB_MAIL =
PAIR1_EXT = _R1
PAIR2_EXT = _R2
BOWTIE2_IDX_PATH = /home/lixingze/XHS-Analysis/HiC-Pro/reads #比对的reads文件目录
BOWTIE2_GLOBAL_OPTIONS = --very-sensitive -L 30 --score-min L,-0.6,-0.2 --end-to-end --reorder
BOWTIE2_LOCAL_OPTIONS = --very-sensitive -L 20 --score-min L,-0.6,-0.2 --end-to-end --reorder
GENOME_SIZE = /home/lixingze/XHS-Analysis/HiC-Pro/reads/genome.sizes #genome.sizes的绝对路径
## Digestion Hi-C
GENOME_FRAGMENT = /home/lixingze/XHS-Analysis/HiC-Pro/reads/genome_dpnii.bed #绝对路径
LIGATION_SITE = GATCGATC #限制性内切酶,具体用的什么酶可以咨询测序公司,我这里用的dnp II
MIN_FRAG_SIZE = 100
MAX_FRAG_SIZE = 100000
MIN_INSERT_SIZE = 100
MAX_INSERT_SIZE = 1000
## Contact Maps
BIN_SIZE = 20000 40000 150000 500000 1000000 #根据自身需求设置 bin size
MATRIX_FORMAT = upper
结果
hic_result/matrix目录
data:存放validpair及其他无效数据文件matrix:存放不同分辨率矩阵文件,
分为raw和iced文件,raw:原始矩阵iced:ice校正后的矩阵后续分析使用,
可以使用HiCPlotter、HiCExplorer出图,或者进行三维基因组学中的部分分析。
pic:存放统计结果图片
stats:存放统计表
hic_result/data目录
allVaildPairs:合并后的valid pairs数据
DEPairs: Dangling end pairs数据
DumpPairs:实际片段长度和理论片段长度不同的数据REPairs:酶切片段重新连接的pairs
FiltPairs:基于min/max insert/fragment size过滤的pairsSCPairs:片段自连的pairs
hic_result/pic目录
plotHiCContactRanges_Example1.pdf有效互作中各类型比例图
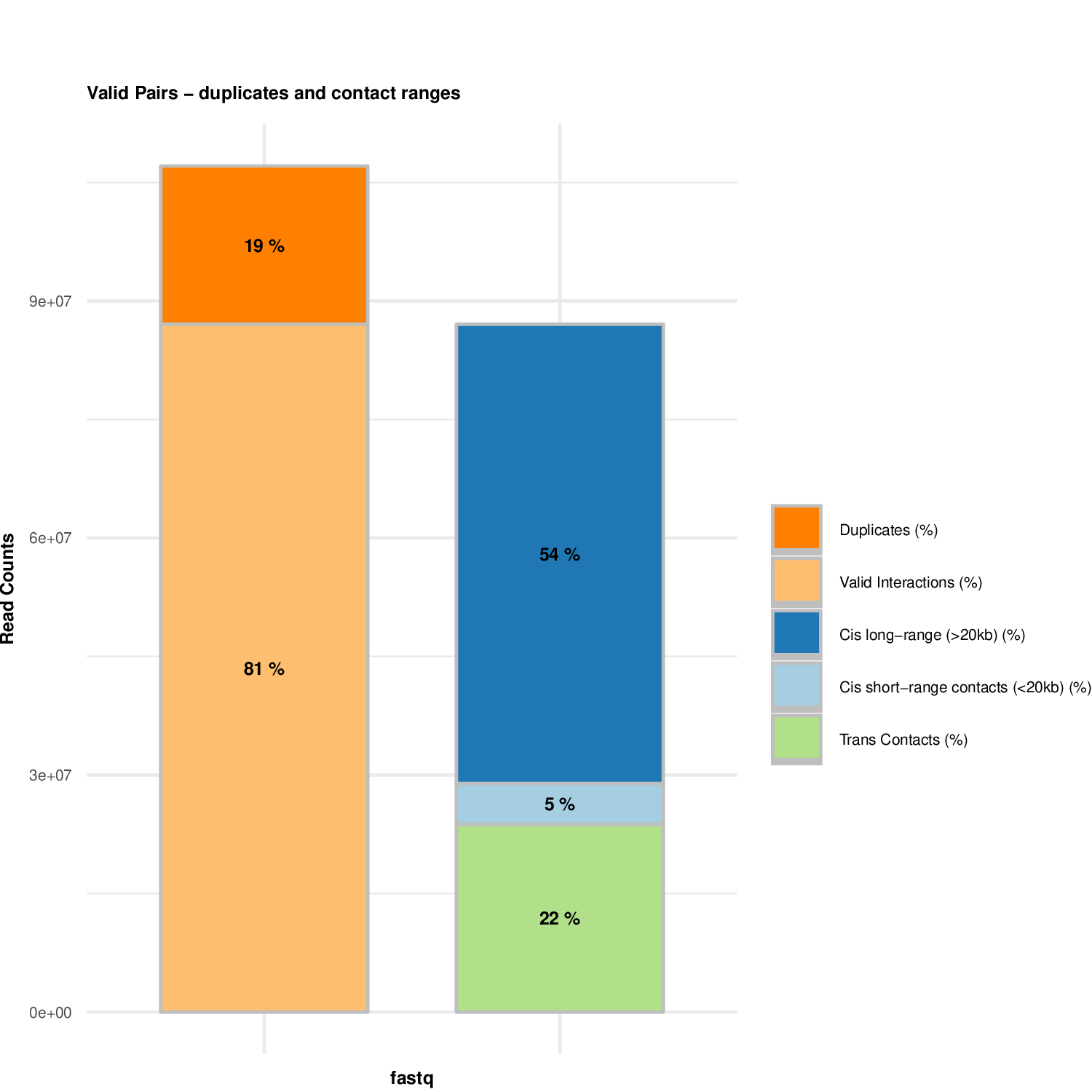
plotHiCFragmentSize_Example1.pdf有效互作的片段大小分布图

plotMappingPairing_Example1.pd合并后双端比对过滤结果图
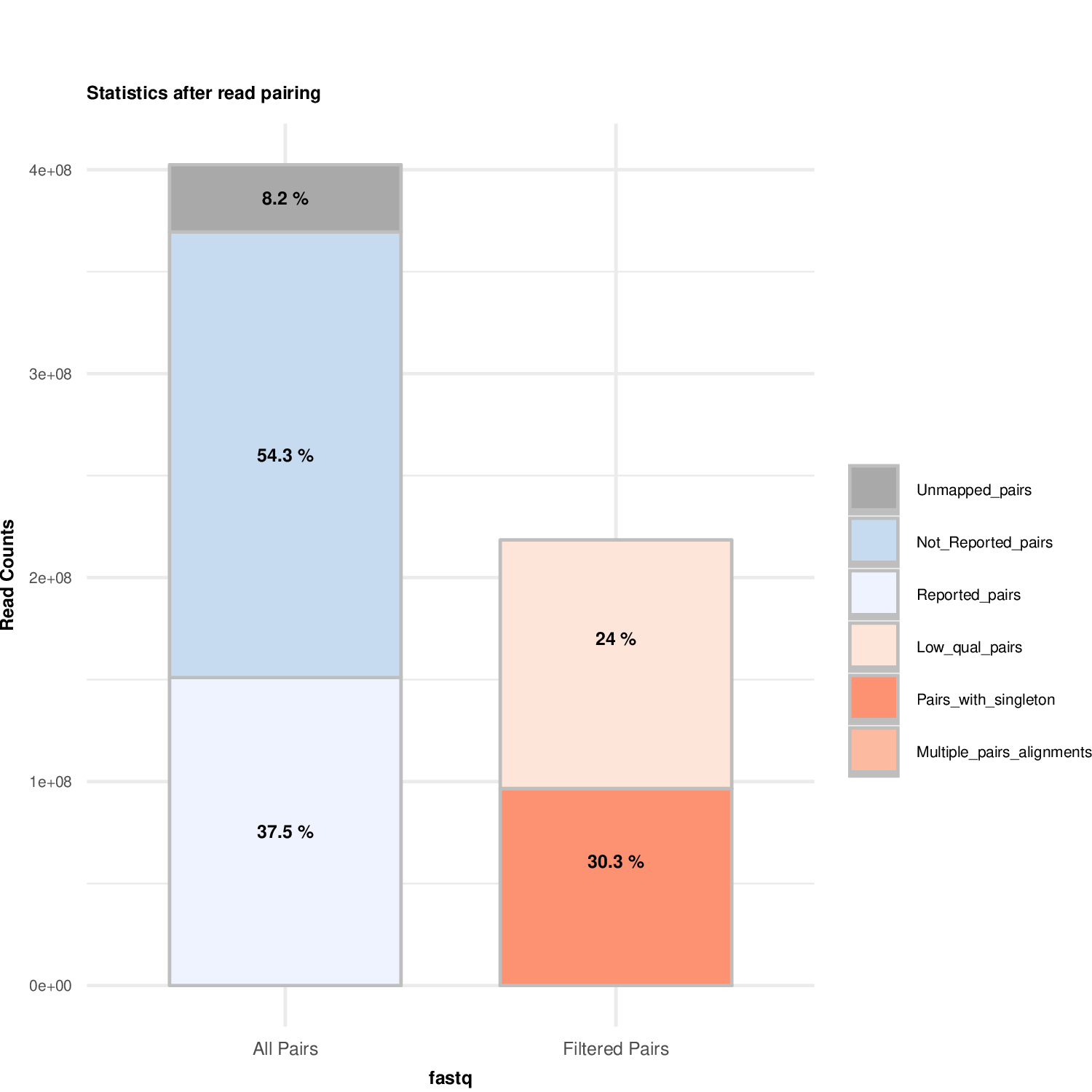
plotHiCFragment_Example1.pdf有效数据过滤结果图

plotMapping_Example1.pdf单端比对过滤结果图
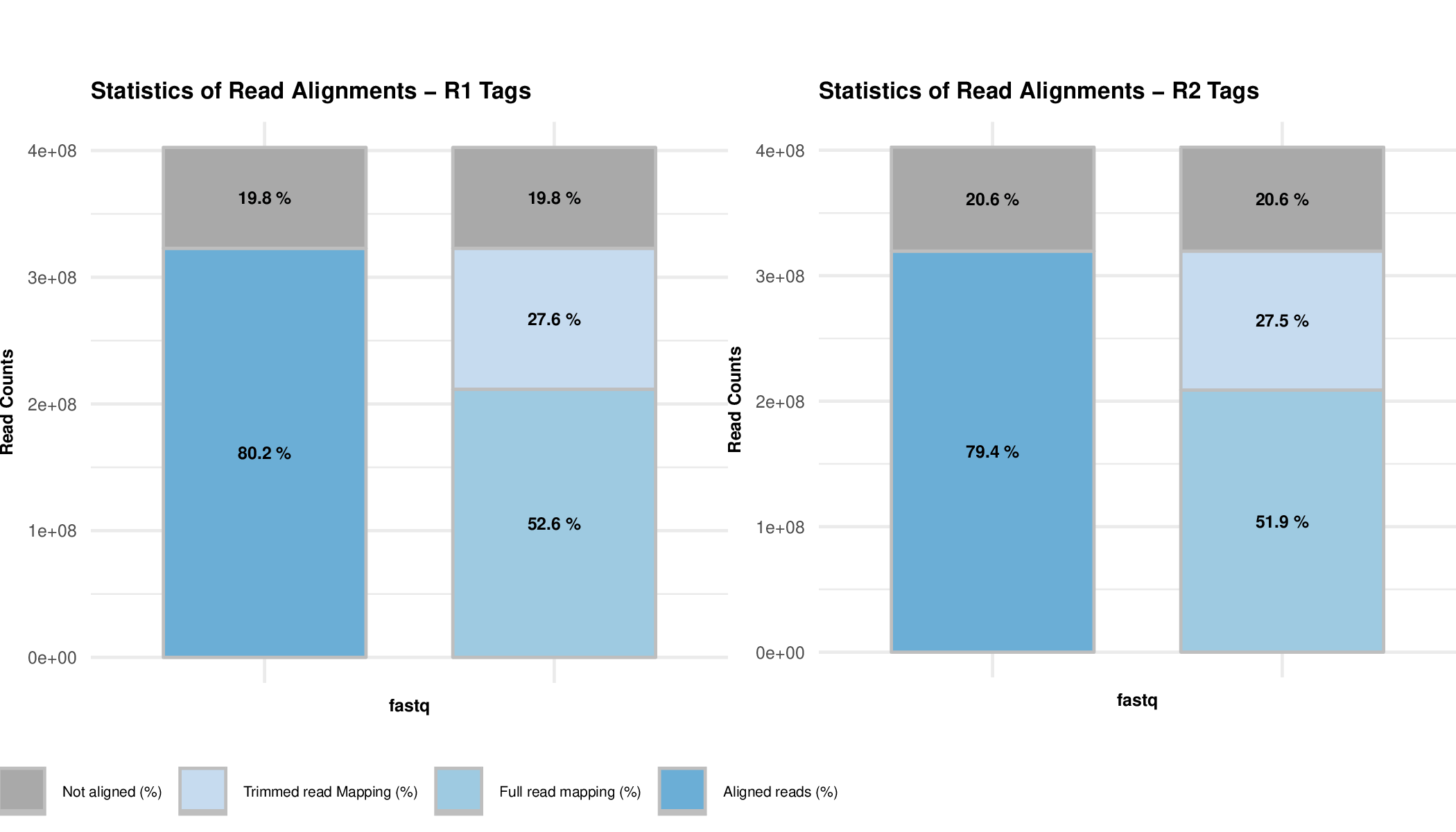
HiC-Pro易报错的地方总结
错误一
Exit: Error: Directory Hierarchy of rawdata '/home/lixingze/data/HiC/hicpro/data' is not correct. No '.fastq(.gz)' files detected
整理reads目录结构
注意:这里在HiCPro的源码中只会读入指定目录的子目录的文件 ,所以将hic测序数据放在子目录下即可
错误二
Pairing of R1 and R2 tags ...
Logs: logs/fastq/mergeSAM.log
make: *** [/home/lixingze/software/HiC-Pro-3.0.0/bin/../scripts//Makefile:144: bowtie_pairing] Error 1
原因可能是之前的bowtie2索引数据有问题造成的,重新跑一次。
总结
不同的报错内容很大程度是个人和环境的原因,所以因人而异,不具有普适性
















 801
801











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









