







 JMC特刊探讨人工智能在药物发现领域的应用,包括分子表征、模型解释、推荐系统、反应设计及生成模型,展示AI如何推动化学信息学与药物化学的进步。
JMC特刊探讨人工智能在药物发现领域的应用,包括分子表征、模型解释、推荐系统、反应设计及生成模型,展示AI如何推动化学信息学与药物化学的进步。
JMC推出 "Artificial Intelligence in Drug Discovery "特刊,强调人工智能(AI)在制药研究中的新兴作用。本期的一个焦点是阐明人工智能方法如何开始影响药物发现的实践。特刊包含了从不同角度看待AI在药物发现中的文章和观点。

本期的论文涵盖了各种方法开发工作和实际应用,让研究者了解到人工智能是如何进入药物发现领域的。当然,机器学习方法已经在化学信息学和计算药物化学中应用了二十多年,但深度学习最近已经成为包括化学在内的许多科学领域的热门话题。
+
分子表征
机器学习方法创建了一个模型,用于建立输入数据和可观察终点之间的关系。药物化学中通常对化学结构和物理性质或生物活性之间的关系进行建模。这个过程中,一个关键的组成部分是用于将分子结构映射成可由机器学习算法处理的形式表示。分子通常被表示为编码分子中结构模式子结构的向量。近年来,一些小组开发了使用深度学习来创建新的 "表示学习 "的方法。虽然这些学习表征的预测能力仍然是一个未知数,但它们已经显示出有希望的初步结果。
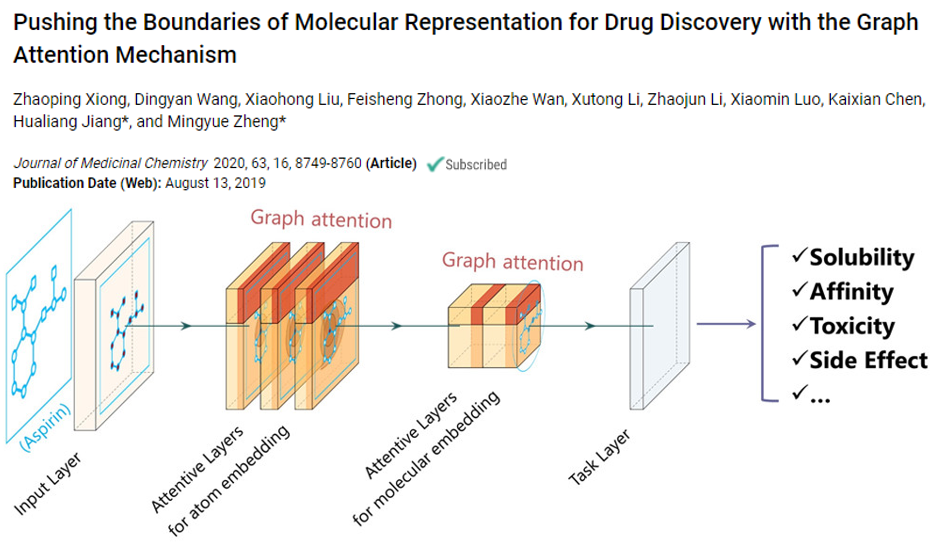
推荐阅读
+
模型解释
许多机器学习方法的缺点之一是它们大多是“黑匣子预测器(black box predictors)”。药物发现环境中,输入化学结构并接收结果,而无需任何解释如何或为什么生成预测。理想情况下,研究人员希望拥有可以由人类用户解释的机器学习模型。模型解释有两个目的:首先,用户可以检查解释,确认它与理论和实验基础相符,并对预测建立一定的信心。其次,对该模型的解释可以为正在被建模的生物活动背后的机理提供线索,并为新分子的设计提供灵感。
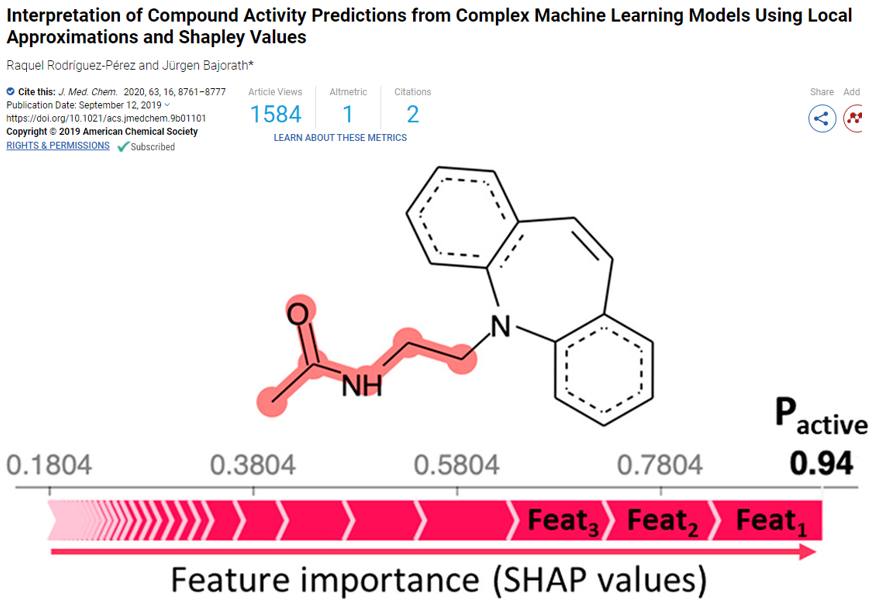
RaquelRodríguezPérez, Jürgen Bajorath. Interpretation of Compound Activity Predictions from Complex Machine Learning Models Using Local Approximations and Shapley Values[J]. Journal of Medicinal Chemistry, 2019.
推荐阅读
Nat. Mach. Intell.|从局部解释到全局理解的树模型
NAACL | 通过对抗性修改,探究链接预测的鲁棒性和可解释性
+
推荐系统
提供建议的计算机系统已成为人们日常生活的一部分。例如,电子商务网站会根据人们的购买历史记录提供建议。在线流媒体网站会推荐人们可能喜欢的音乐和视频。这一概念扩展到了药物化学实验室,如推荐有机合成路线,相似化合物的三维结构以及可能提供更多见解的测定方法。
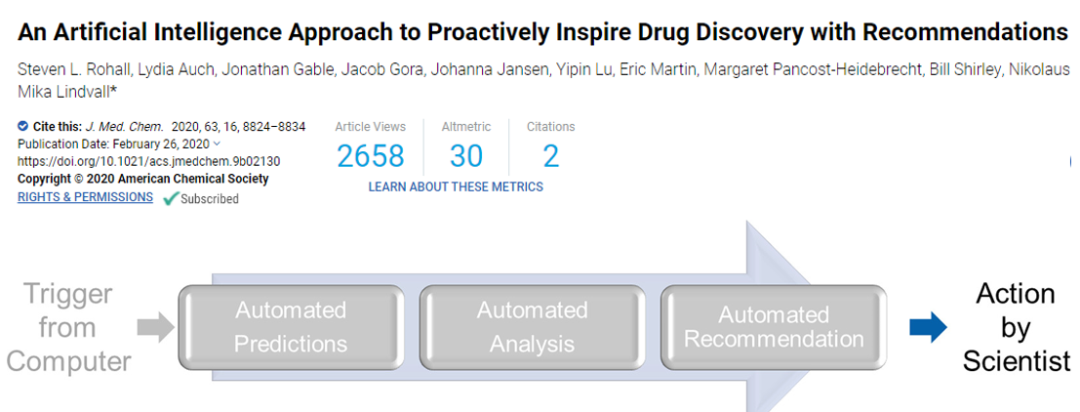
An Artificial Intelligence Approach to Proactively Inspire Drug Discovery with Recommendations. Steven L. Rohall, Lydia Auch, Jonathan Gable, Jacob Gora, Johanna Jansen, Yipin Lu, Eric Martin, Margaret Pancost-Heidebrecht, Bill Shirley, Nikolaus Stiefl, and Mika Lindvall. Journal of Medicinal Chemistry 2020 63 (16), 8824-8834
DOI: 10.1021/acs.jmedchem.9b02130
+
反应设计
AI目前正在取得进展的化学领域之一是预测和建模新的化学反应和合成路线。
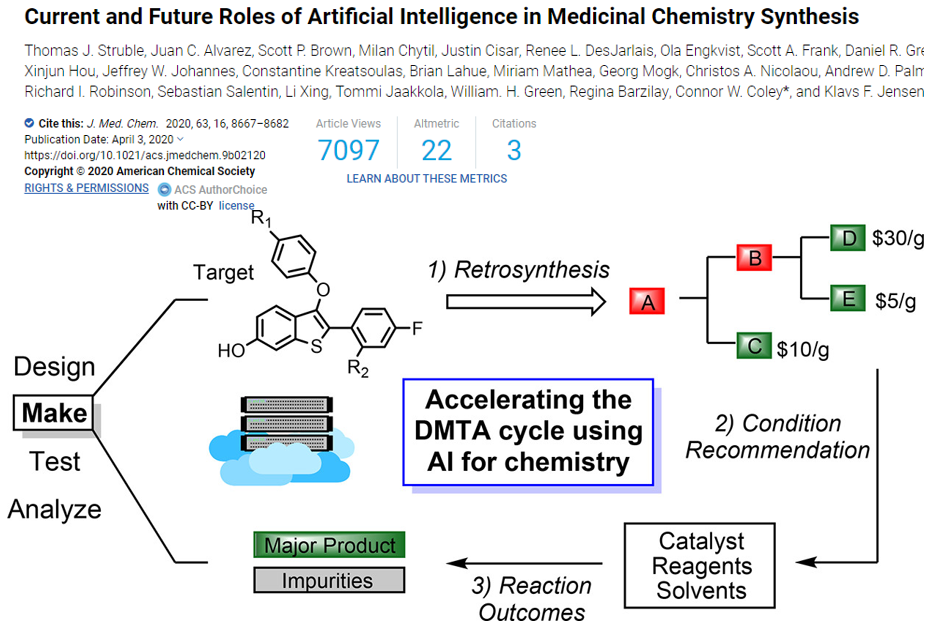
推荐阅读
ICML2020 | Retro*: 通过神经引导的A*搜索学习逆合成设计
ICML2020 | G2Gs:不依赖模板的的逆合成预测新框架
+
生成模型
尽管计算化学取得了30多年的进步,但在药物发现中,许多新分子的构想也源于药物化学家的想象力和独创性。从1990年开始,许多小组创建了用于进行从头分子设计的计算机程序。这些程序通常通过在蛋白质结合位点的背景下“生长”现有分子来运行。但是,从头设计获得了一些成功的故事,但该技术未能获得主流采用。过去的几年中,研究人员看到了一种称为生成建模技术的兴起。这个领域起源于语言模型和图像生成,它以一组分子结构作为输入,这些分子结构被编码为连续的低维表示。然后可以对该表示进行解码以生成新颖的分子。但是,化学家可能会质疑这种系统学习生成药物样分子所必需的实际化学反应的能力。
推荐阅读
J. Cheminform. | 基于SMILES的利用骨架的分子生成模型
Chem. Sci. | SyntaLinker: 基于Transformer神经网络的片段连接生成器
J. Cheminform.| Mol-CycleGAN:基于Graph的分子生成优化模型
Nat. Mach. Intell. | 利用条件循环神经网络生成特定性质分子
Nat. Mach. Intell. | 少量数据的生成式分子设计
基于生成式深度学习方法设计潜在2019-nCoV蛋白酶抑制剂
参考资料
Artificial Intelligence in Drug Discovery: Into the Great Wide Open. Jürgen Bajorath, Steven Kearnes, W. Patrick Walters, Nicholas A. Meanwell, Gunda I. Georg, and Shaomeng Wang Journal of Medicinal Chemistry 2020 63 (16), 8651-8652.
DOI: 10.1021/acs.jmedchem.0c01077


















 2629
2629

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











