






蛋白质分子量
蛋白质是由许多氨基酸残基通过肽键(一个氨基酸的 α-羧基与另一个氨基酸的 α-氨基脱水缩合形成的化学键)连接而成。蛋白质的分子量(molecular weight)为各个氨基酸的分子量之和,是蛋白质的重要理化参数。单位为Dalton(道尔顿,缩写Da,或D),定义为碳12原子质量的1/12,1D=1/N g,N为阿弗加德罗常数。通常,我们可以使用氨基酸残基数*110大概计算蛋白质的分子量。
目录
1)UniProt数据库
打开https://www.uniprot.org,在输入框输入“P05130”,然后点击“Search”按钮。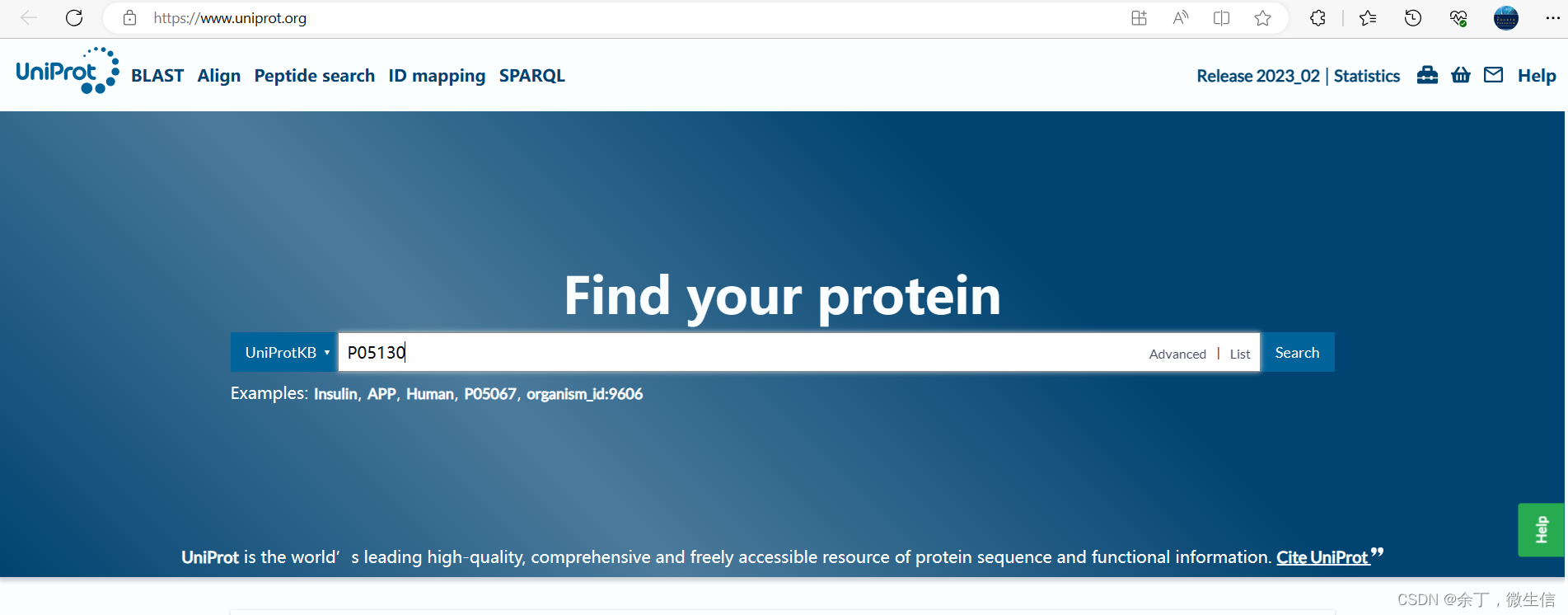
点击结果页面左侧的“Sequence & Isoform”,链接到序列处。此处有长度679,质量77,695 Da。粗略等于679*110。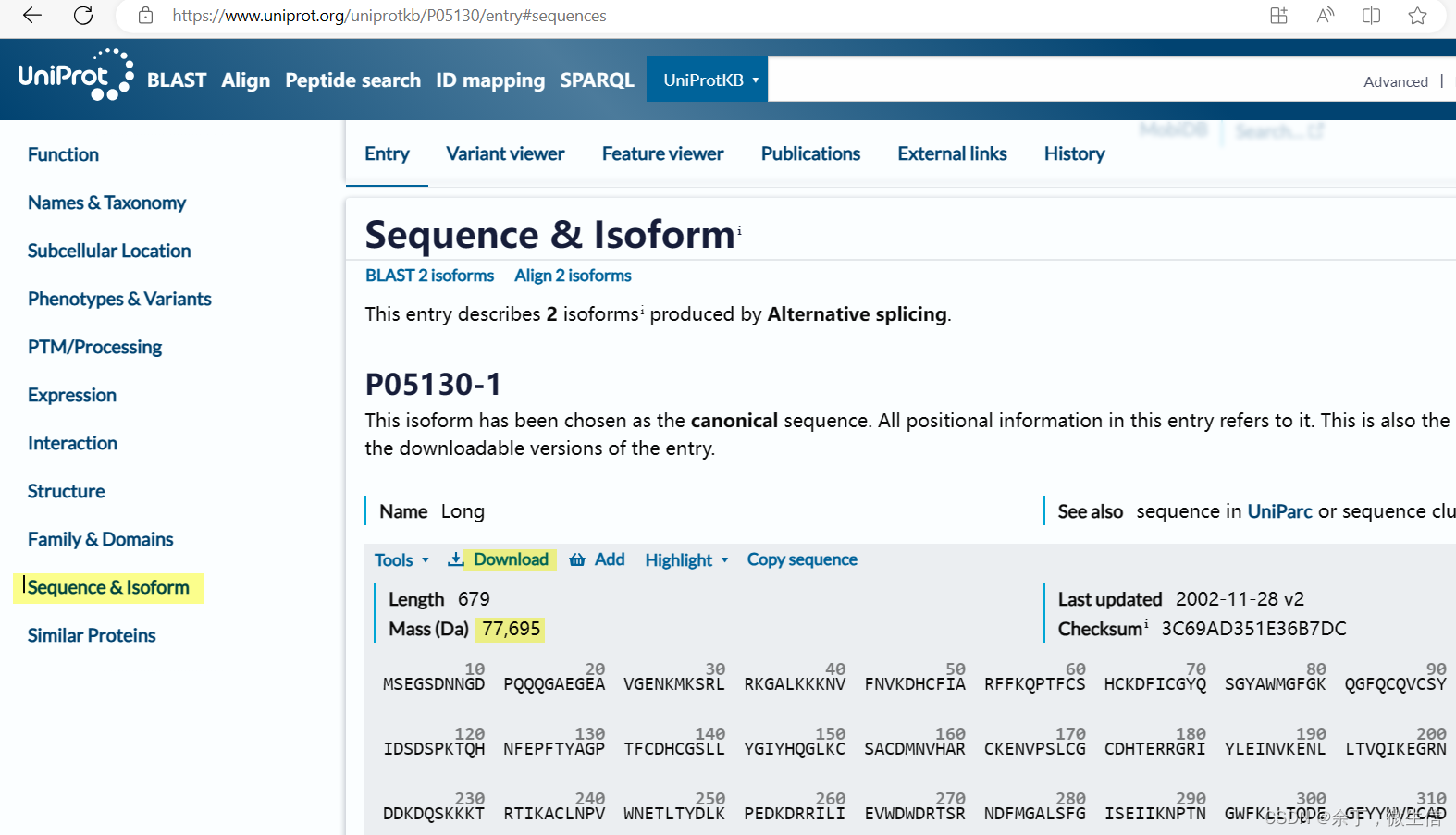
点击“Download”下载序列备用。
如果仅有几条蛋白质,并且有对应的UniProtKB id,建议直接检索获得结果。
如果有N条蛋白质,并且有对应的UniProtKB id,可以使用UniProt的idmapping工具检索,批量下载结果,然后进行解析以获得分子量。
2)Expasy
打开Expasy - ProtParam tool,在上面的输入框中输入蛋白质id或者序列id,例如P05130或者KPC1_DROME;或者在下面的输入框中输入蛋白质单字母序列。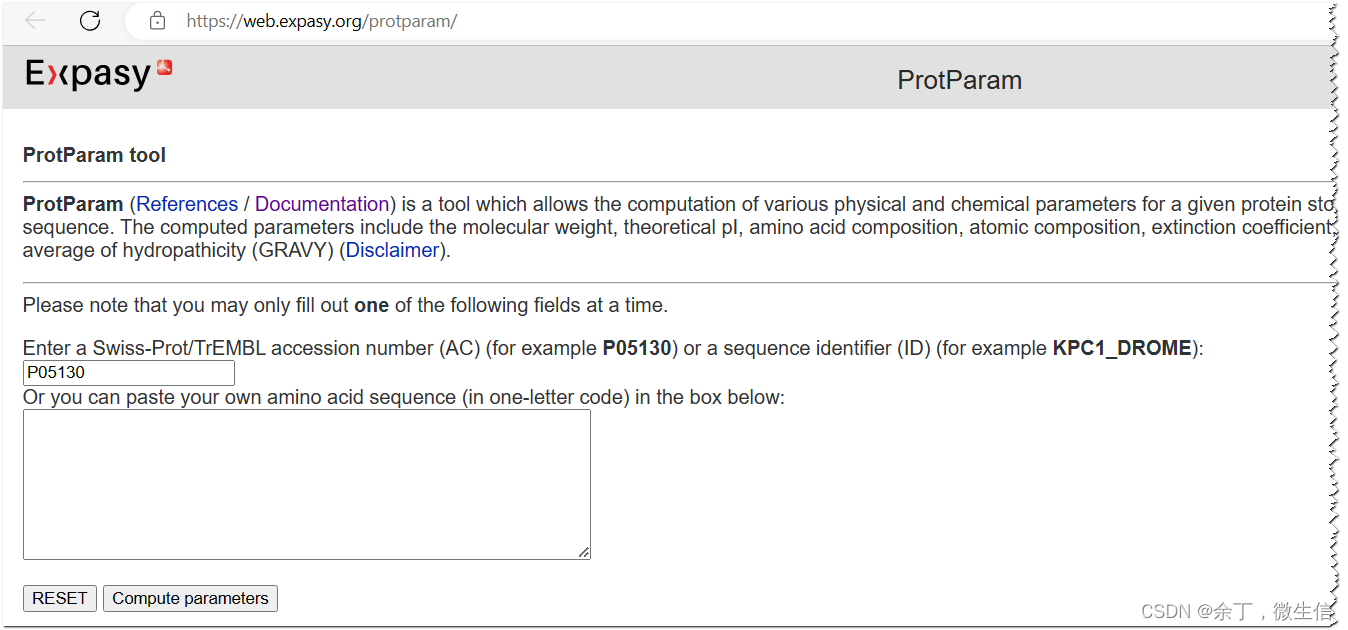
点击“compute parameters”提交,弹出来参数,全部默认,点击“Submit”按钮。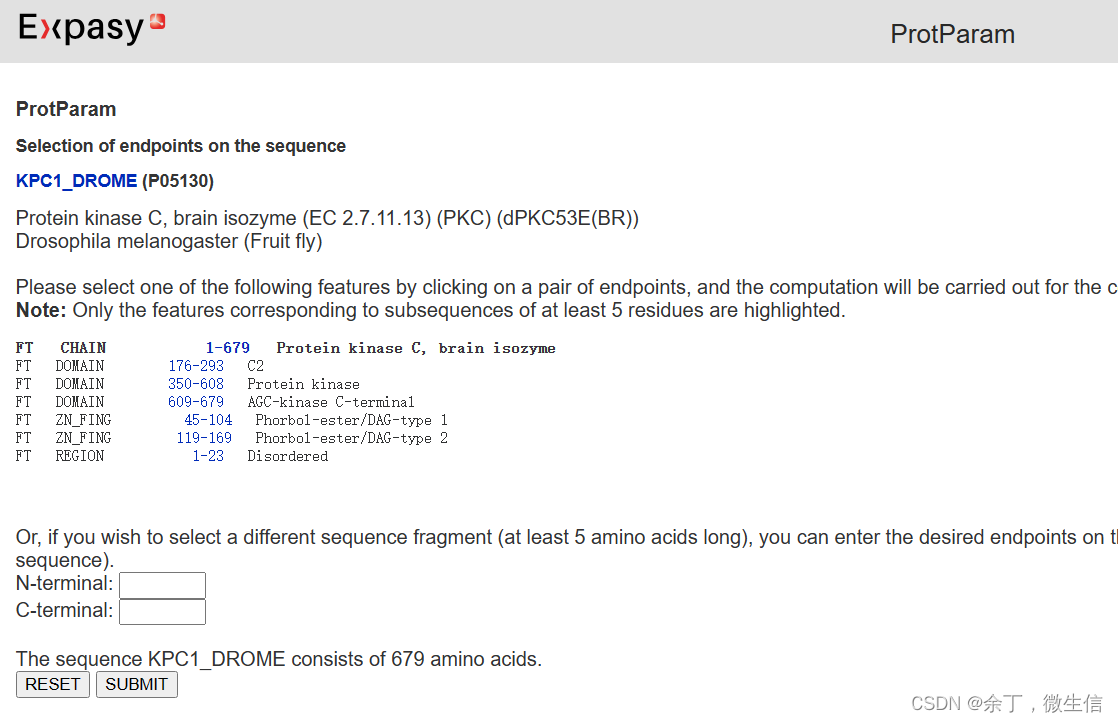
分子量就计算出来了。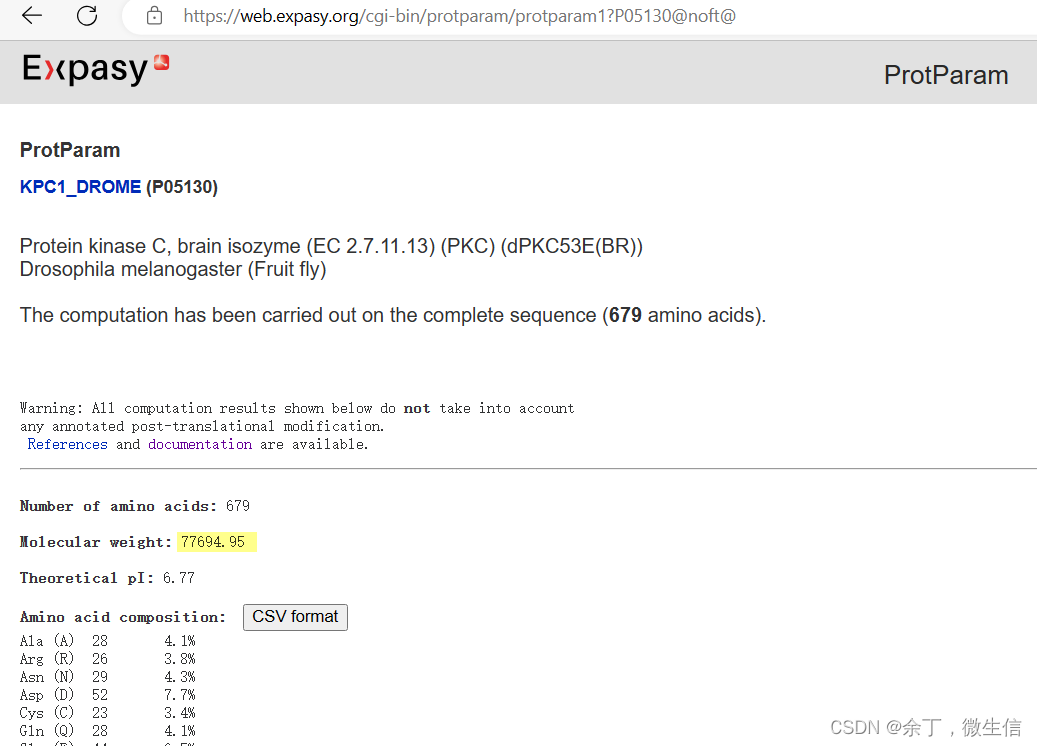
也可以输入我们在UniProt上获得的序列(这里仅序列)计算蛋白质分子量。略,请大家自行尝试。
3)EMBOSS
打开https://www.ebi.ac.uk/Tools/seqstats/emboss_pepstats/,在输入框中输入在UniProtKB上获得的序列(fasta格式),点击“Submit”按钮提交。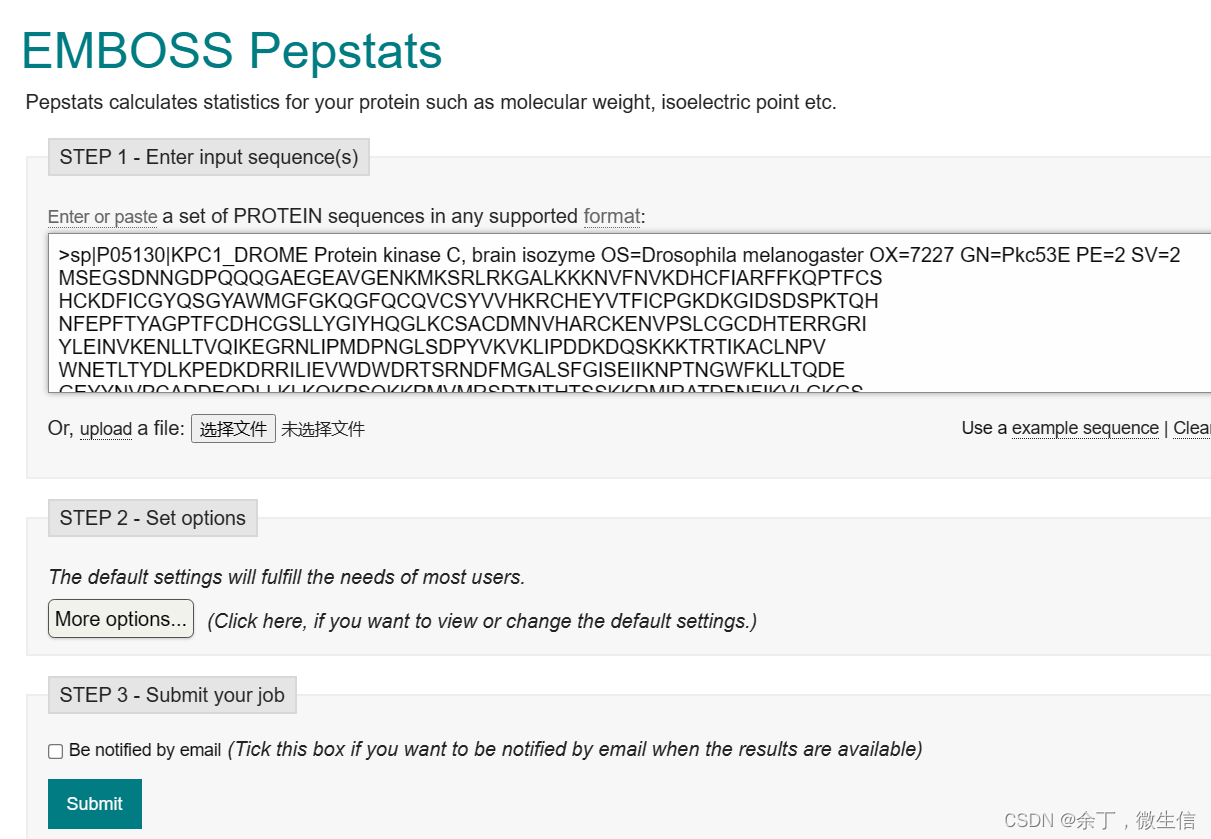
经过约10秒后,会返回结果。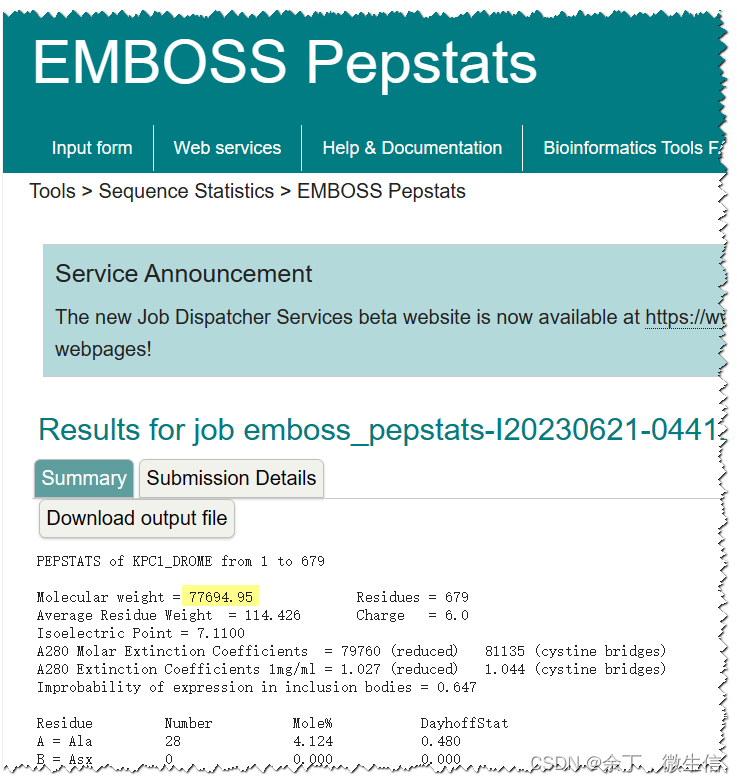
也可以一次性提交N条序列(fasta格式)。略,请大家自行尝试。
4)PIR
打开Composition/Molecular Weight Calculation [PIR - Protein Information Resource],在上面的输入框输入UniProtid或者在下面的输入框中输入在UniProt上获得的序列,点击“Submit”按钮。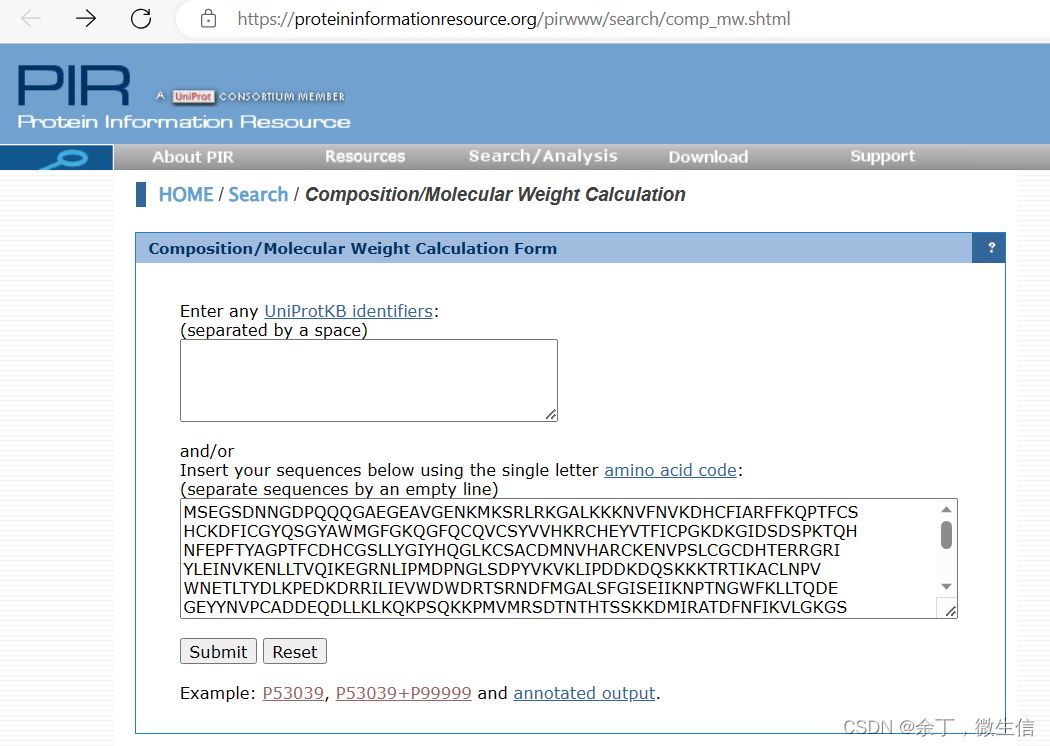
约1秒后返回结果。红框中为计算所用的公式和每个氨基酸的分子量(带水分子的分子量)。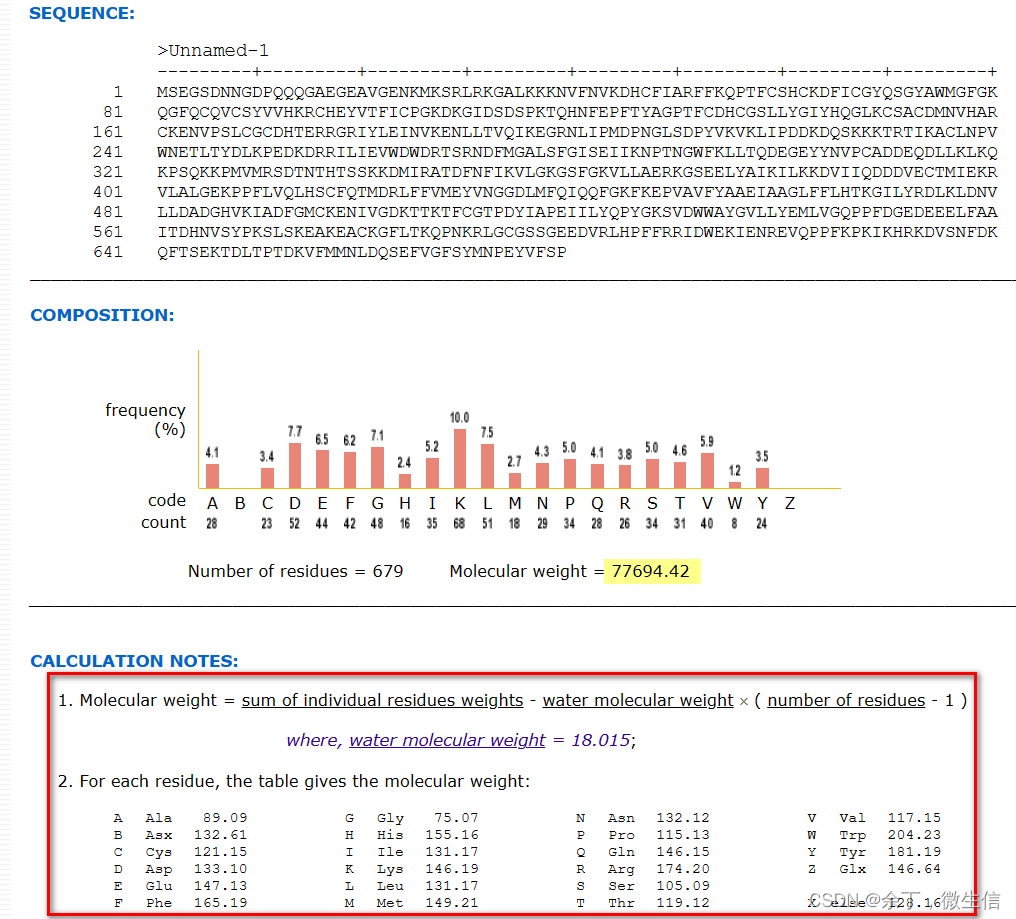
也可以一次性提交N条序列(换行分割)。略,请大家自行尝试。
总结:
| 工具 | 输入 | 批量 | 新序列 | 速度 | 批量结果 |
| Uniprot | UniProtKB id | Yes | NO | 快 | 下载后解析 |
| Expasy | UniProtKB id/sequence | NO | Yes | 快 | |
| EMBOSS | sequence | Yes | Yes | 慢 | 解析网页 |
| PIR | UniProtKB id/sequence | Yes | Yes | 快 | 解析网页 |
二、python代码版
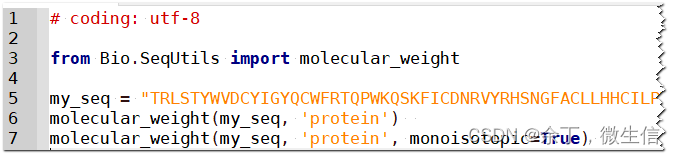
2)python代码从头计算
虽然Biopython 3行代码就可以搞定蛋白质分子量计算,但是我们需要知道计算原理。最重要的就是获得氨基酸对应的分子量表格。
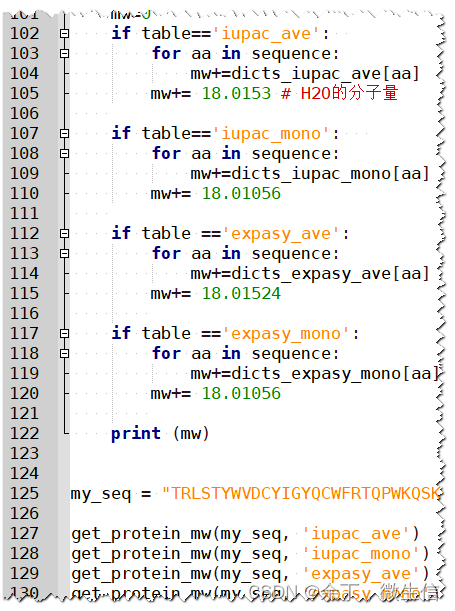
代码解释:
输入1:单字母的氨基酸序列
输入2:每个氨基酸对应的分子量表格
原理:根据每个氨基酸对应的分子量表格,将所有字母对应的分子量数值加起来,然后再加上水分子的分子量,就是最终的分子量。
注意:这里的分子量是不带水分子的,如果你用的表格是带水分子的,需要用PIR结果中的计算公式。
总结:
1)网站上使用的基本都是平均质量。
2)使用上述6种方法计算出来的P05130的分子量基本一样,小数点细微差别可能是由于精度不同或者使用的分子量表格(见附2)不同。
| 工具 | P05130分子质量 |
| UniProt | 77695 |
| Expasy | 77694.95 |
| EMBOSS | 77694.95 |
| PIR | 77694.42 |
| Biopython-IUPAC | 77694.0959 |
| Python-Expasy | 77694.94634 |
附1:Average vs monoisoform
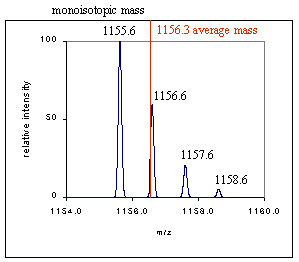
蛋白质/化合物的平均质量(average mass,红线处),是由其组成的元素质量的加和,每种元素的质量选取所有同位素的平均质量。
蛋白质/化合物的单同位素质量(monoisoform,最高峰处),是由其组成元素质量的加和,每种元素质量选择其最大丰度同位素的质量。
绝大多数情况下,平均质量>单同位素质量(见附2)。
附2:氨基酸分子量表格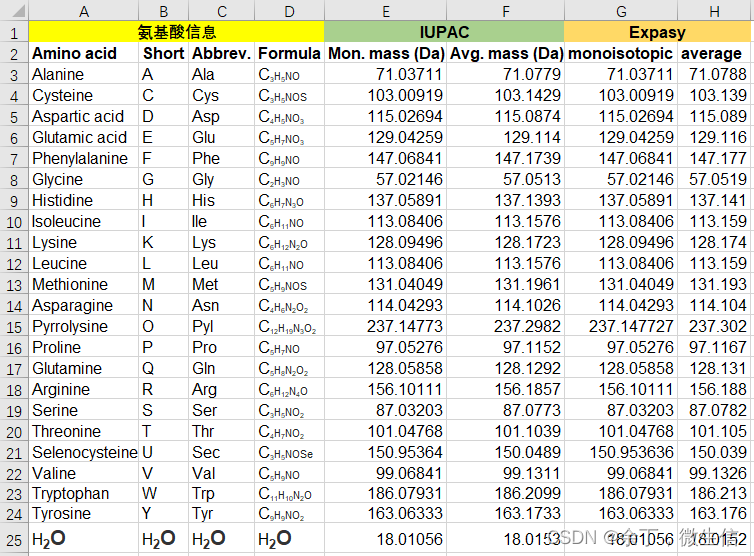
参考:
[1] molecular_weight in SeqUtils · Issue #3859 · biopython/biopython · GitHub
[2] X.molecular_weight() · Issue #4002 · biopython/biopython · GitHub
[3] https://en.wikipedia.org/wiki/Proteinogenic_amino_acid#Mass_spectrometry
微生信助力高分文章,用户175000+,谷歌学术3200

















 473
473

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









