






基因本体论(Gene Ontology,GO)计划(http://www.geneontology.org)为注释基因、基因产物和序列开发了一套结构化的、受控词汇表。它被分成三部分:分子功能(Molecular Function,MF)、生物过程(Biological Process,BP)和细胞组分(Cell Component,CC)。
GO功能富集分析结果,一般都会包含至少4列:GO term,基因数,P值和类别。今天给大家带来一张非常直观,颜值也颇高的GO富集结果可视化图。
1,打开绘图页面
首先,使用浏览器(推荐chrome或者edge)打开GO BP、CC、MF三合一双侧条形图绘制页面,左侧为常见作图导航,中间为数据输入框和可选参数,右侧为描述和结果示例。
http://www.bioinformatics.com.cn/plot_basic_GO_term_bp_cc_mf_left_right_bar_plot_191
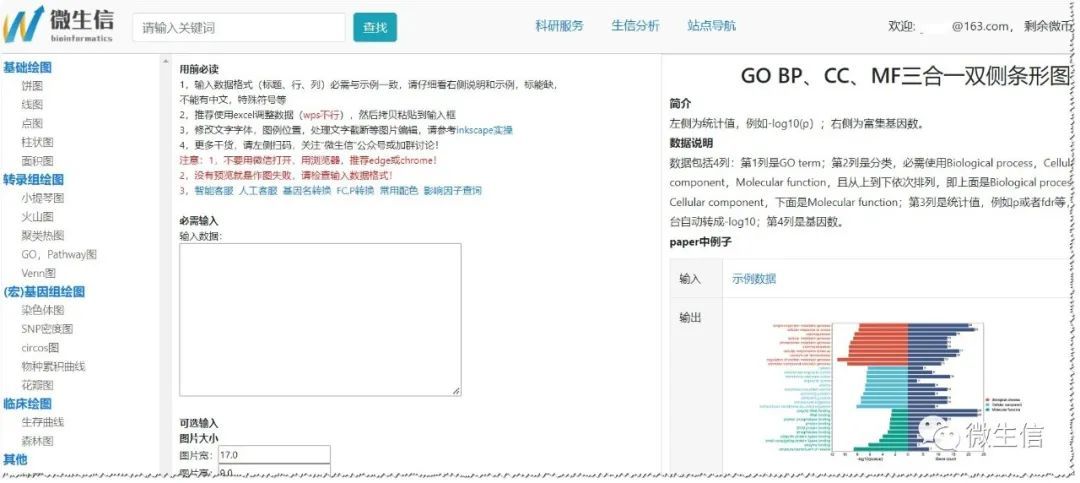
图1.可视化页面
2,示例数据
点击右侧“示例数据”链接下载excel格式的示例数据。
示例数据(仅供参考)包括4列:
第1列是GO term名字;
第2列是分类(分类名必需是Biological process、Cellular component、Molecular function,且按照顺序排列);
第3列是Pvalue(或者FDR、qvalue等,程序会默认自动将其转化成-log10值,因此p值不能为0),每个类别之内的条目可以按照P值从小到大,或者从大到小排列;
第4列是基因数;
注意:你需要参考示例数据,将自己的富集结果在excel中整理成示例数据的样式。
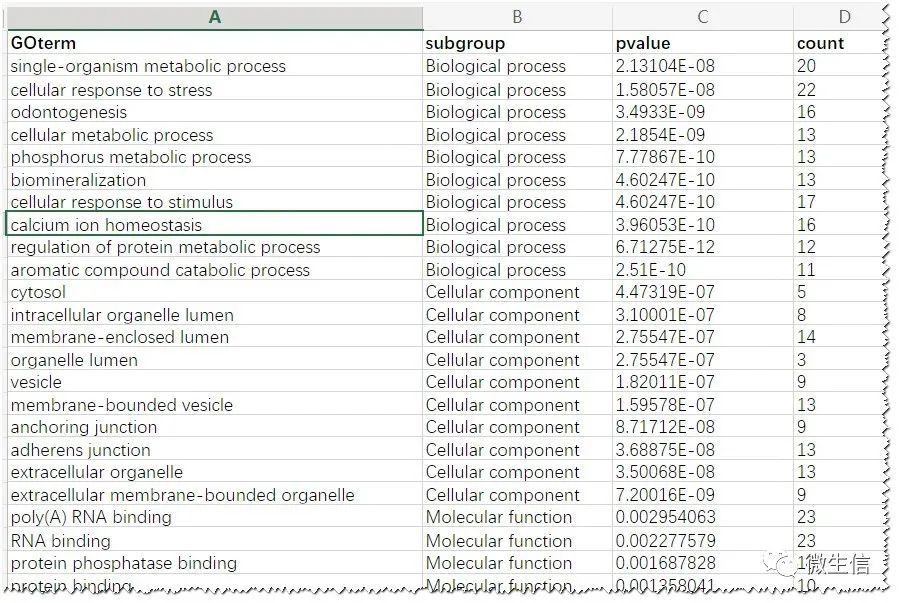
图2. 输入数据示例
3,粘贴示例数据
直接复制示例数据中的ABCD 4列数据,然后粘贴到输入框。
注意:不是拷贝excel文件,是拷贝excel文件里边的数据哦。另外粘贴到输入框后,格式乱了没关系,只要在excel中是整齐的就行。并且数据矩阵中不能有空的单元格,中文字符等。

图3. 必需输入
4,修改参数,并提交
我们设置了图片尺寸,颜色等参数,基本能满足日常绘图使用。如需更高级的定制,请联系我们。
部分参数详解:
颜色:这里我们还是使用Nature Publishing Group的配色。
左侧最大值:用来控制左侧的柱子距离左边界的距离,请使用可整除的整数
右侧最大值:用来控制右侧的柱子距离右边界的距离,请使用可整除的整数
这两个值需要与图片长宽配合着使用,以保证零点位置不是0.0。

图4. 颜色等可调参数
5,提交出图
输入数据粘贴好,参数调整好后,点击提交按钮,约3秒后,在页面右侧会出现结果预览图。我们提供了4种图片格式供下载使用,两种矢量图(pdf,svg)和两种标量图(600 dpi tiff和300 dpi png)。
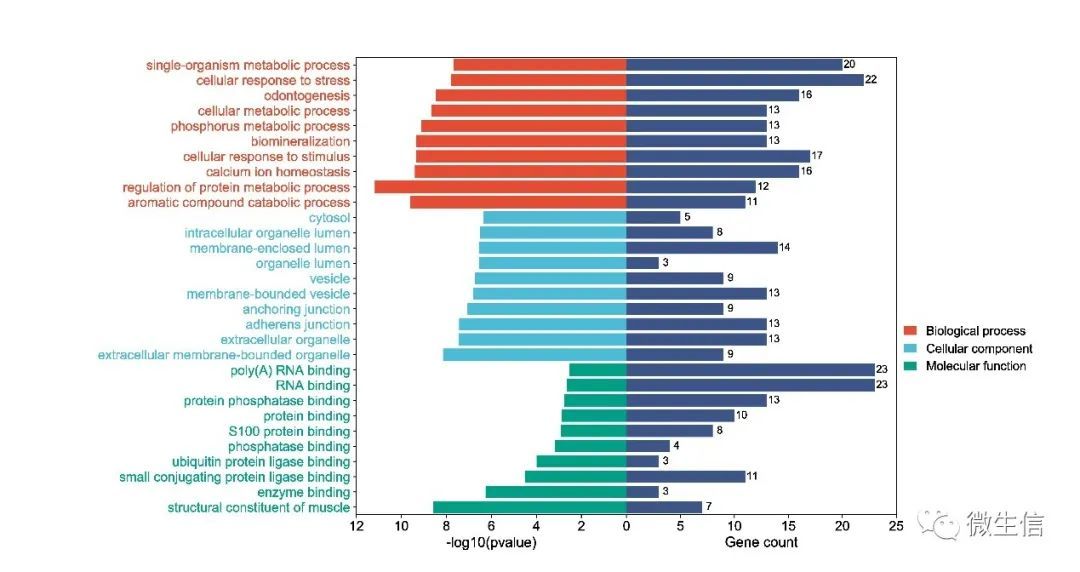
图左侧表示GO term的统计p值(-log10转化),p值越小,条形越长;并且按照颜色区分三部分;
图右侧表示富集的基因数,基因数越多,条形越长。
点评:与气泡图相比,该图的优点在于一眼就能分辨出那个term的基因数最多,P值最小。因为人们对于颜色的分辨程度要低于对尺寸的分辨程度,特别是在颜色差别不大的情况下。
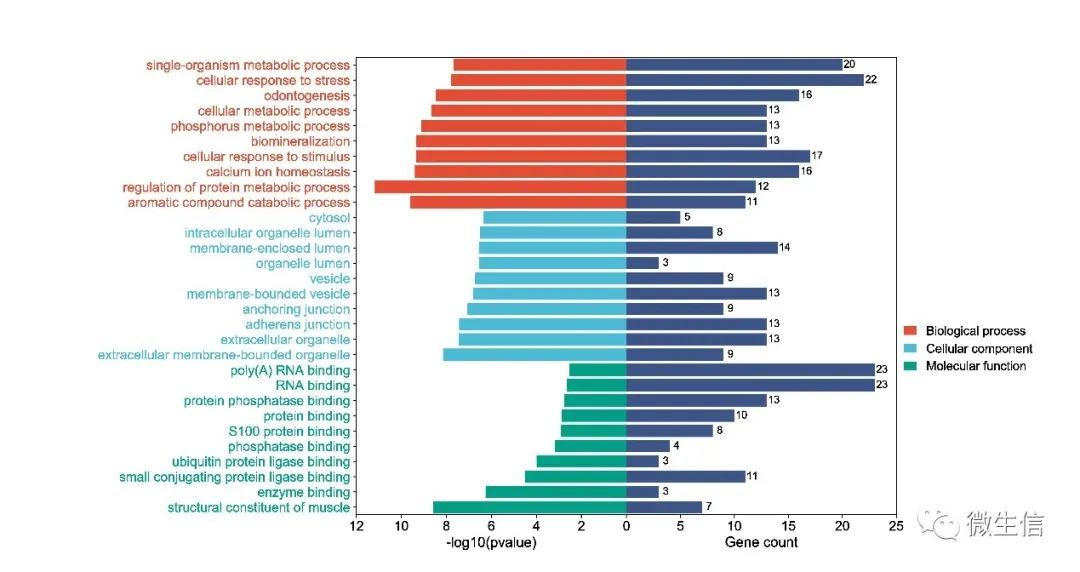
图5.预览与下载
没有预览就是没有出图,这时请参考示例数据,检查输入数据格式。
遇到文字截断,需要修改字体、调整字体大小等,使用scape软件。

















 529
529

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









