







前言
这部分内容继续介绍没介绍完的生物学常用格式:
- BED
- Sam
- Bam
- VCF
BED
BED(Browser Extensible Data)格式也是一种常用的用于基因注释的数据格式。BED格式数据有专门开发的工具bedtools来专门处理,和之前提到的GFF文件有很多相似的地方。
BED数据格式如下;
chr7 127471196 127472363 Pos1 0 + 127471196 127472363 255,0,0
chr7 127472363 127473530 Pos2 0 + 127472363 127473530 255,0,0
chr7 127473530 127474697 Pos3 0 + 127473530 127474697 255,0,0
chr7 127474697 127475864 Pos4 0 + 127474697 127475864 255,0,0
chr7 127475864 127477031 Neg1 0 - 127475864 127477031 0,0,255
chr7 127477031 127478198 Neg2 0 - 127477031 127478198 0,0,255
chr7 127478198 127479365 Neg3 0 - 127478198 127479365 0,0,255
chr7 127479365 127480532 Pos5 0 + 127479365 127480532 255,0,0
chr7 127480532 127481699 Neg4 0 - 127480532 127481699 0,0,255BED文件每行至少包括chrom,chromStart,chromEnd三列(必选);另外还可以添加额外的9列(可选)
必选的三列:
- chrom:染色体或者是scaffold的名称,或scaffold是比染色体小一点的组装单位。
- chromStart:染色体或scaffold中特征的起始位置。通常第一个碱基编号为0。
- chromEnd:染色体或scaffold中特征的结束位置。
9个可选的BED字段:
- name:定义每一行属性的名称
- score:得分在0到1000之间,如果useScore参数为1,则得分是指灰度级别
- strand:表示链的方向,+表示正向,或者-表示负向
- thickStart: 起始位置,例如,基因显示中的起始密码子
- thickEnd:终止位置,例如:基因终止编码位置
- itemRgb:RGB形式的RGB值,例如(255,255,255)。但是需要 itemRgb属性设置为On
- blockCount:BED行中的外显子数目
- blockSizes: 用逗号分割的外显子的大小, 对应于blockCount的数目
- blockStarts:用逗号分割的列表, 所有外显子的起始位置,数目也与blockCount数目对应
Sam
SAM(sequence alignment map)是一种序列比对的文件格式,Sam格式是高通量测序数据分析的常用格式,因为它可以快速查找与坐标重叠的比对(它有记录一些序列索引信息),并且是一种高效的存储方式,例如Bam就是Sam格式的二进制文件,是一种通用的格式。同时Sam格式也是是目前最常用的存放比对或联配数据的格式。无论是重测序,还是转录组,还是表观组,几乎所有流程都会产生SAM/BAM文件作为中间步骤,用于接下来分析。
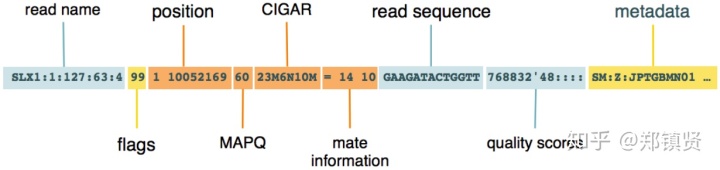
Sam样例格式如下:

Sam格式数据可以分为两部分,分别是注释信息(header section)和比对结果部分(alignment section),注释信息可有可无,都是以@开头,用不同的tag表示不同的信息,主要有以下几种格式:
- @HD,说明符合标准的版本、对比序列的排列顺序
- @SQ,参考序列说明
- @RG,比对上的序列(read)说明
- @PG,使用的程序说明
- @CO,任意的说明信息
比对结果部分,每一行表示一个片段的比对信息,包括11个顺序固定的必选字段和一个可选的字段,字段之间用tag分割。这11个字段包括:
- QNAME:比对片段的(read)的名称信息
- FLAG:位标识,表明比对类型:paring,strand,mate strand等等
- RNAME:比对上的参考序列名
- POS,position缩写,read比对到参考序列上,第一个碱基所在的位置,没匹配上设置为0
- MAPQ:Mapping quality缩写,比对的质量分数,越高说明位置越唯一且可信度越高
- CIGAR:(Compact Idiosyncratic Gapped Alignment Report),简要比对信息表达式,其以参考序列为基础,使用数字加字母表示比对结果,比对结果信息,匹配碱基数,可变剪接等等
- RNEXT:下一个片段比对上的参考序列的编号,没有另外的片段,这里是*,同一个片段,用=**
- PNEXT:下一个片段比对上的位置,如果不可用,此处为0
- TLEN:观察到的Template的长度,最左边得为正,最右边的为负,中间的不用定义正负,不可用为0
- SEQ:序列片段的序列信息,如果不存储此类信息设置为*,
- QUAL:序列的质量信息,格式同FASTQ一样,read质量的ASCII编码
可选字段(optional fields),格式如:TAG:TYPE:VALUE,其中TAG有两个大写字母组成,每个TAG代表一类信息,每一行一个TAG只能出现一次,TYPE表示TAG对应值的类型,可以是字符串、整数、字节、数组等:
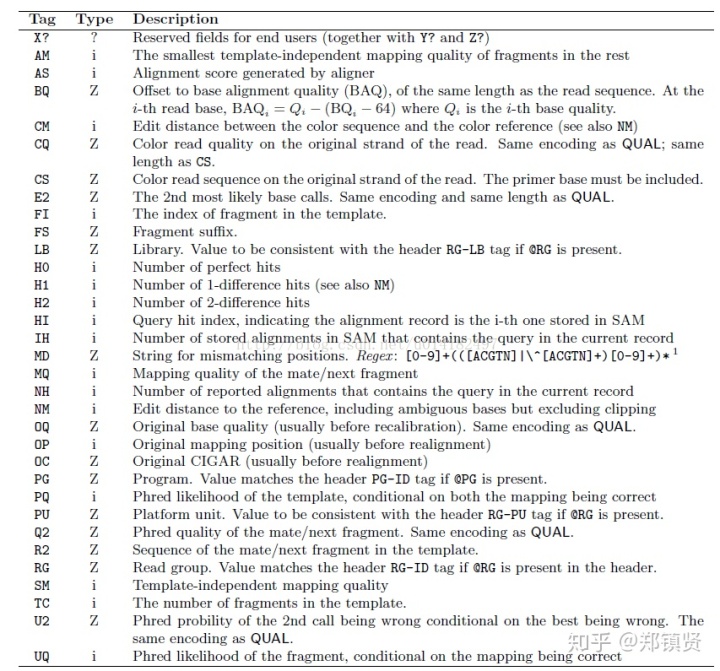
为了方便大家理解,从网上找了个图:
Bam
Bam是Sam的二进制文件格式,Bam格式中的b是binary的意思,由于Sam格式文件大小通常是十分巨大的,所以为了减少存储量等因素而将Sam转换为二进制格式以便于分析
VCF
VCF(Variant Call Format)是用于描述SNP,INDEL和SV结果的文本文件,是存储变异位点的标准格式
以下是VCF格式的一个样例:
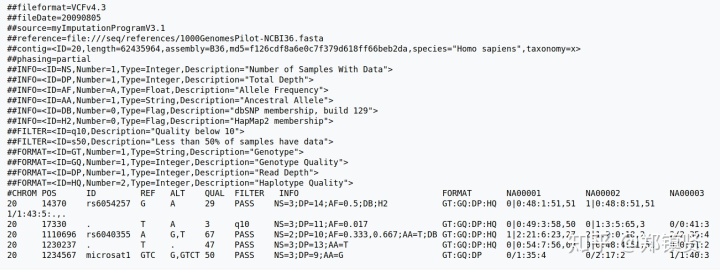
这个数据可以包括两个部分:
- 注释部分:##表示,可以找到接下来每一个位点的描述的tag
- 主体部分:包含10列数据,主题部分每一行代表一个variant的信息
分别是:
- CHROM : 参考序列名称
- POS : variant所在的位置,如果是INDEL的话,位置是INDEL的第一个碱基位置
- ID : variant的ID。同时对应着dbSNP数据库中的ID,若没有,则默认使用.
- REF : 参考序列的碱基
- ALT : variant的碱基
- QUAL : variants的质量。Phred格式的数值,代表着此位点是纯合的概率,此值越大,则概率越低,代表着次位点是variants的可能性越大(表示变异碱基的可能性)
- FILTER : 用于表示次位点是否要被过滤掉
- INFO : variant的相关信息,这里有很多的内容
- FORMAT : variants的格式和基因型的信息
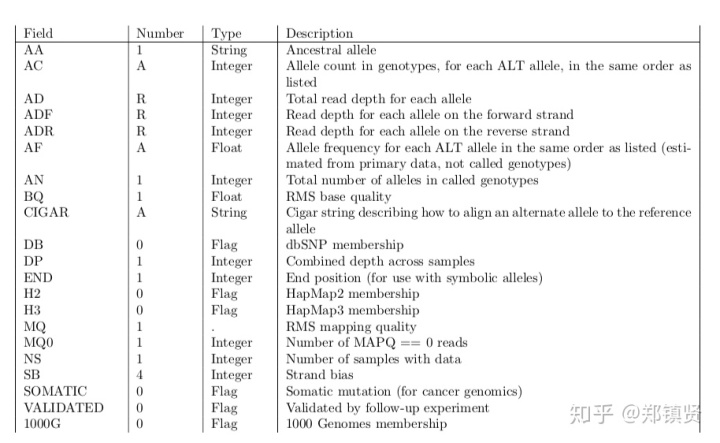
VCF的INFO
上面有一列的数据是INFO的列名,这里包含了很多其他信息,大家可以参考wiki的内容查看:
本次生物信息学数据格式介绍介绍部分就到这,欢迎和我讨论!
欢迎大家关注我的知乎专栏:从零开始生物信息学
相同内容也可以关注我的微信公众号: 壹读基因:















 573
573











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









