






 本文详细介绍了VCF文件的组成结构,包括元信息、头部、数据行以及标记信息,解释了各部分的作用和常见格式。同时,文章提供了一些处理VCF文件的工具,并附上相关链接供读者深入学习。
本文详细介绍了VCF文件的组成结构,包括元信息、头部、数据行以及标记信息,解释了各部分的作用和常见格式。同时,文章提供了一些处理VCF文件的工具,并附上相关链接供读者深入学习。
Introduction
VCF is a text file format (most likely stored in a compressed manner). It contains meta-information lines, a header line, and then data lines each containing information about a position in the genome. The format also has the ability to contain genotype information on samples for each position.
Example VCF file
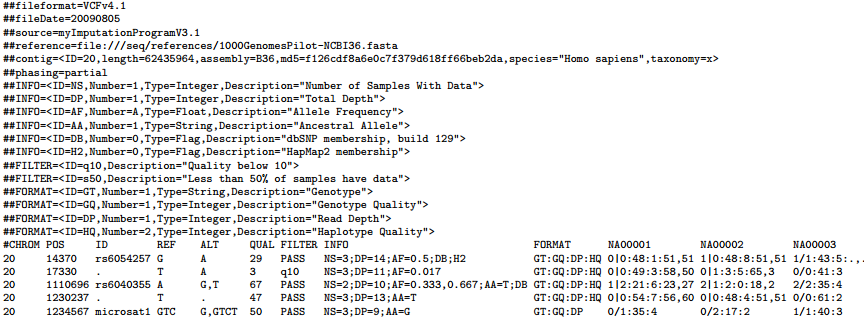
Meta-information lines
File meta-information is included after the ## string and must be key=value pairs. It is strongly encouraged that information lines describing the INFO, FILTER and FORMAT entries used in the body of the VCF file be included in the meta-information section. Although they are optional, if these lines are present then they must be completely well-formed.
A single ‘fileformat’ field is always required, must be the first line in the file, and details the VCF format version number. For example, for VCF version 4.1, this line should read:
##fileformat=VCFv4.1
INFO fields should be described as follows (all keys are required):
##INFO=<ID=ID,Number=number,Type=type,Description="description">
FILTERs that have been applied to the data should be described as follows:
##FILTER=<ID=ID,Description="description">
Likewise, Genotype fields specified in the FORMAT field should be described as follows:
##FORMAT=<ID=ID,Number=number,Type=type,Description="description">
Possible Types for FORMAT fields are: Integer, Float, Character, and String (this field is otherwise defined precisely as the INFO field).
Marker information
There are 8 fixed fields per record. All data lines are tab-delimited. In all cases, missing values are specified with a dot (‘.’). Fixed fields are:
1. CHROM - chromosome: An identifier from the reference genome
2. POS - position: The reference position, with the 1st base having position 1.
3. ID - identifier: Semi-colon separated list of unique identifiers where available. If this is a dbSNP variant it is encouraged to use the rs number(s).
4. REF - reference base(s): Each base must be one of A,C,G,T,N (case insensitive). Multiple bases are permitted.
5. ALT - alternate base(s): Comma separated list of alternate non-reference alleles called on at least one of the samples.
6. QUAL - quality: Phred-scaled quality score for the assertion made in ALT. High QUAL scores indicate high confidence calls.
7. FILTER - filter status: PASS if this position has passed all filters. Otherwise,if the site has not passed all filters, a semicolon-separated list of codes for filters that fail.
8. INFO - additional information: INFO fields are encoded as a semicolon-separated series of short keys with optional values in the format: <key>=<data>[,data]. Arbitrary keys are permitted, although the following sub-fields are reserved (albeit optional):
• AA : ancestral allele
• AC : allele count in genotypes, for each ALT allele, in the same order as listed
• AF : allele frequency for each ALT allele in the same order as listed: use this when estimated from primary
data, not called genotypes
• AN : total number of alleles in called genotypes
• BQ : RMS base quality at this position
• CIGAR : cigar string describing how to align an alternate allele to the reference allele
• DB : dbSNP membership
• DP : combined depth across samples, e.g. DP=154
• END : end position of the variant described in this record (for use with symbolic alleles)
• H2 : membership in hapmap2
• H3 : membership in hapmap3
• MQ : RMS mapping quality, e.g. MQ=52
• MQ0 : Number of MAPQ == 0 reads covering this record
• NS : Number of samples with data
• SB : strand bias at this position
• SOMATIC : indicates that the record is a somatic mutation, for cancer genomics
• VALIDATED : validated by follow-up experiment
• 1000G : membership in 1000 Genomes
The exact format of each INFO sub-field should be specified in the meta-information (as described above).
The first sub-field must always be the genotype (GT) if it is present. There are no required sub-fields.
As with the INFO field, there are several common, reserved keywords that are standards across the community:
• GT : genotype, encoded as allele values separated by either of / or |. The allele values are 0 for the reference allele (what is in the REF field), 1 for the first allele listed in ALT, 2 for the second allele list in ALT and
so on. For diploid calls examples could be 0/1, 1 | 0, or 1/2, etc. For haploid calls, e.g. on Y, male nonpseudoautosomal X, or mitochondrion, only one allele value should be given; a triploid call might look like 0/0/1. If a call cannot be made for a sample at a given locus, ‘.’ should be specified for each missing allele in the GT field (for example ‘./.’ for a diploid genotype and ‘.’ for haploid genotype). The meanings of the separators are as follows (see the PS field below for more details on incorporating phasing information into the genotypes):
◦ / : genotype unphased
◦ | : genotype phased
• DP : read depth at this position for this sample (Integer)
• FT : sample genotype filter indicating if this genotype was “called” (similar in concept to the FILTER field).
• GL : genotype likelihoods comprised of comma separated floating point log10-scaled likelihoods for all possible genotypes given the set of alleles defined in the REF and ALT fields.
• GLE : genotype likelihoods of heterogeneous ploidy, used in presence of uncertain copy number.
• PL : the phred-scaled genotype likelihoods rounded to the closest integer (and otherwise defined precisely as the GL field) (Integers)
• GP : the phred-scaled genotype posterior probabilities (and otherwise defined precisely as the GL field); intended to store imputed genotype probabilities (Floats)
• GQ : conditional genotype quality, encoded as a phred quality −10log10p(genotype call is wrong, conditioned on the site’s being variant) (Integer)
• HQ : haplotype qualities, two comma separated phred qualities (Integers)
• PS : phase set.
• PQ : phasing quality, the phred-scaled probability that alleles are ordered incorrectly in a heterozygote (against all other members in the phase set).
• EC : comma separated list of expected alternate allele counts for each alternate allele in the same order as listed in the ALT field (typically used in association analyses) (Integers)
• MQ : RMS mapping quality, similar to the version in the INFO field. (Integer)
FORMAT:
GT指genotype, 1/1 代表homozygote, 两个等位基因均和参考基因组不同。 0/1 代表heterozygote, 一个等位基因与参考基因组一样,另一个不一样。
DP:read depth at this postion for this sample
RO:和reference一样的碱基有多少个 QR : RO的质量
AO:和reference不一样的碱基多少个 QA : AO的质量
GL : 三种基因型的likelihood,越接近于0,可能性越大。Sample data
Resources
Here are some tools for manipulating VCF files:
know more about VCF,please click here:
http://www.1000genomes.org/wiki/Analysis/Variant%20Call%20Format/vcf-variant-call-format-version-41

















 2616
2616

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









