






 这是一个Python脚本,用于重新编号PDB文件中的原子和残基。通过命令行参数可以指定输入文件、起始编号,并选择是否重新编号原子和残基。如果指定了--chainreset,则会在遇到新链时重置残基编号。
这是一个Python脚本,用于重新编号PDB文件中的原子和残基。通过命令行参数可以指定输入文件、起始编号,并选择是否重新编号原子和残基。如果指定了--chainreset,则会在遇到新链时重置残基编号。
Python脚本:PDB文件中原子和残基重新编号
Command:
python renumber_pdb.py -i protein.pdb -a -r > output.pdb
renumber_pdb.py
# Python 3 script to atoms and residues in a PDB file.
#
# run
# ./renumber.py -h
# for help
#
class Pdb(object):
""" Object that allows operations with protein files in PDB format. """
def __init__(self, file_cont = [], pdb_code = ""):
self.cont = []
self.atom = []
self.hetatm = []
self.fileloc = ""
if isinstance(file_cont, list):
self.cont = file_cont[:]
elif isinstance(file_cont, str):
try:
with open(file_cont, 'r') as pdb_file:
self.cont = [row.strip() for row in pdb_file.read().split('\n') if row.strip()]
except FileNotFoundError as err:
print(err)
if self.cont:
self.atom = [row for row in self.cont if row.startswith('ATOM')]
self.hetatm = [row for row in self.cont if row.startswith('HETATM')]
self.conect = [row for row in self.cont if row.startswith('CONECT')]
def renumber_atoms(self, start=1):
""" Renumbers atoms in a PDB file. """
out = list()
count = start
for row in self.cont:
if len(row) > 5:
if row.startswith(('ATOM', 'HETATM', 'TER', 'ANISOU')):
num = str(count)
while len(num) < 5:
num = ' ' + num
row = '%s%s%s' %(row[:6], num, row[11:])
count += 1
out.append(row)
return out
def renumber_residues(self, start=1, reset=False):
""" Renumbers residues in a PDB file. """
out = list()
count = start - 1
cur_res = ''
for row in self.cont:
if len(row) > 25:
if row.startswith(('ATOM', 'HETATM', 'TER', 'ANISOU')):
next_res = row[22:27].strip() # account for letters in res., e.g., '1A'
if next_res != cur_res:
count += 1
cur_res = next_res
num = str(count)
while len(num) < 3:
num = ' ' + num
new_row = '%s%s' %(row[:23], num)
while len(new_row) < 29:
new_row += ' '
xcoord = row[30:38].strip()
while len(xcoord) < 9:
xcoord = ' ' + xcoord
row = '%s%s%s' %(new_row, xcoord, row[38:])
if row.startswith('TER') and reset:
count = start - 1
out.append(row)
return out
if __name__ == '__main__':
import argparse
parser = argparse.ArgumentParser(
description='Renumber residues in a pdb file',
formatter_class=argparse.RawTextHelpFormatter
)
parser.add_argument('-i', '--input', help='Input PDB file')
parser.add_argument('-s', '--start', help='Number of the first residue in the renumbered file (default = 1)')
parser.add_argument('-a', '--atoms' ,action='store_true', help='Renumbers atoms')
parser.add_argument('-r', '--residues', action='store_true', help='Renumbers residues')
parser.add_argument('-c', '--chainreset', action='store_true', help='Resets the residue renumbering after encountering a new chain.')
parser.add_argument('-v', '--version', action='version', version='v. 1.0')
args = parser.parse_args()
if not args.input:
print('{0}\nPlease provide an input file.\n{0}'.format(50* '-'))
parser.print_help()
quit()
if not args.start:
start = 1
else:
start = int(args.start)
if not args.atoms and not args.residues:
print('{0}\nPlease provide at least the --atoms or --residues flag.\n{0}'.format(50* '-'))
parser.print_help()
quit()
pdb1 = Pdb(args.input)
if args.atoms:
pdb1.cont = pdb1.renumber_atoms(start=start)
if args.residues:
pdb1.cont = pdb1.renumber_residues(start=start, reset=args.chainreset)
for line in pdb1.cont:
print(line)
效果:
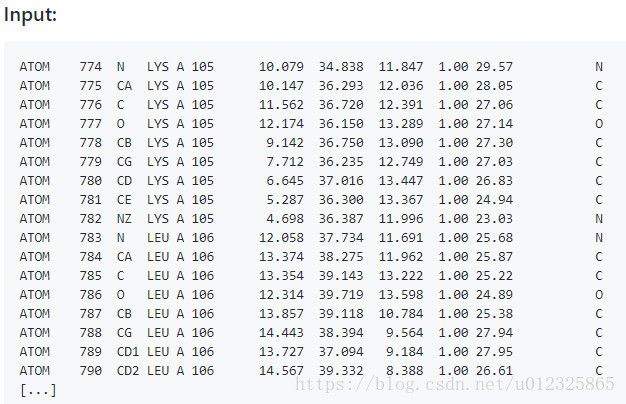
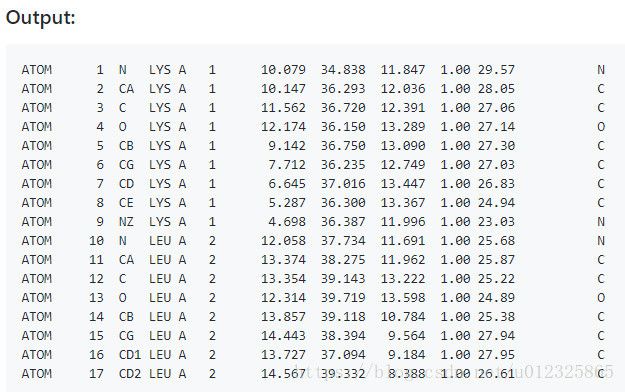
参考:
https://github.com/AspirinCode/protein-science/tree/master/scripts-and-tools/renumber_pdb

















 429
429

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









