






 本文介绍了如何使用Origin和ggplot2软件绘制类似Nature medicine上美观的富集分析折线图。通过Origin的Plot Setup、Plot Details等设置调整散点、线条和标签,以及ggplot2的geom_line、geom_point和geom_segment方法创建图表,再利用ggrepel包解决标签重叠问题,实现专业图表的绘制。
本文介绍了如何使用Origin和ggplot2软件绘制类似Nature medicine上美观的富集分析折线图。通过Origin的Plot Setup、Plot Details等设置调整散点、线条和标签,以及ggplot2的geom_line、geom_point和geom_segment方法创建图表,再利用ggrepel包解决标签重叠问题,实现专业图表的绘制。
实用科研工具推荐 、详实生信软件教程分享、前沿创新组学文章解读、独家生信视频教程发布,欢迎关注微信公众号:基迪奥生物 (gene-denovo)
|本文作者:莫北
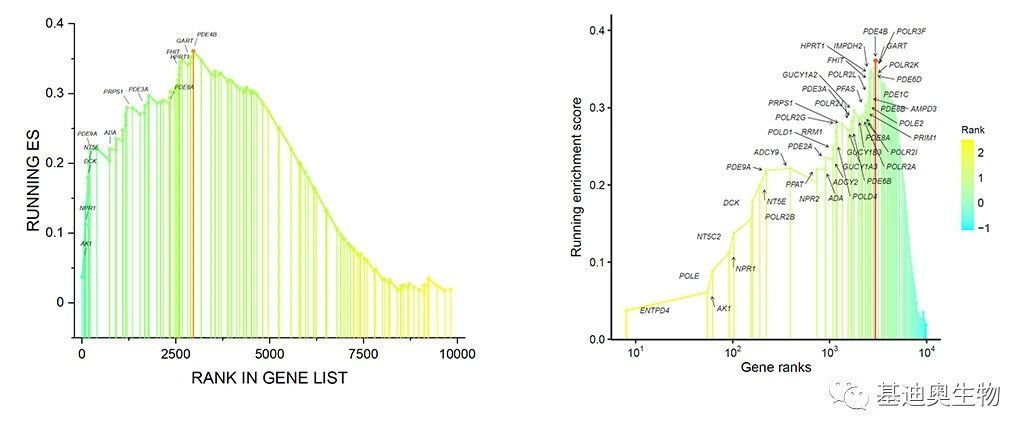
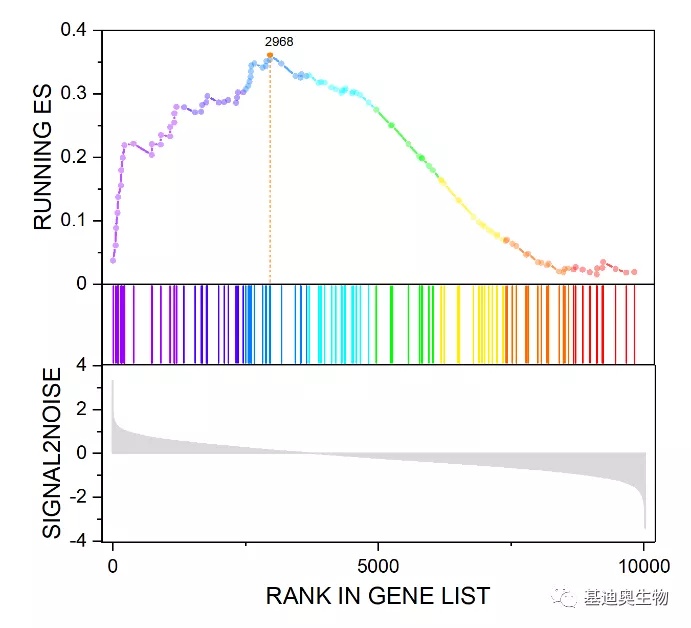
前段时间看到Nature medicine上的一个富集分析折线图(如下),非常漂亮,心仪不已。
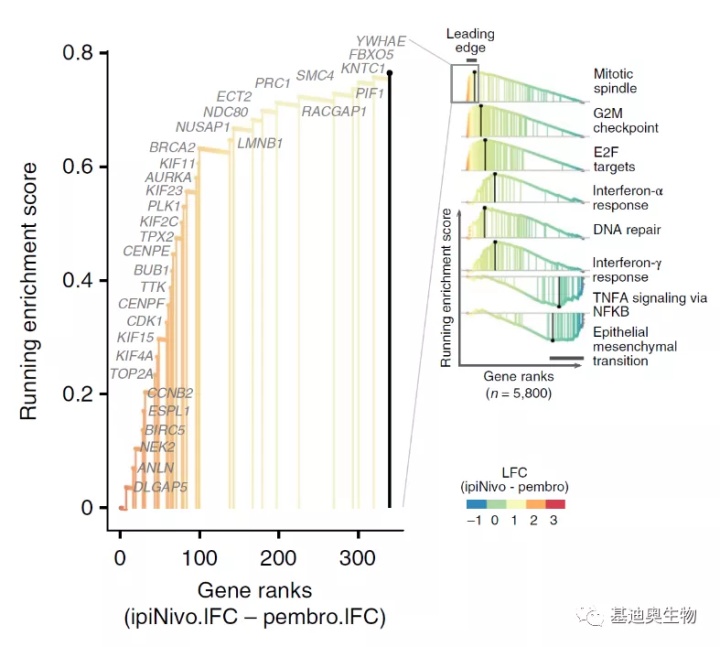
如果直接将基因集富集分析(GSEA)输出结果图放在文章中,就显得非常“粗糙”了,高下立判!那么这样的折线图如何绘制呢?
其实并不难,常规科研作图软件也可以画。闲言少叙,下面就开始介绍具体的画法,内容包括基于Origin、ggplot2的绘图方法和绘图数据准备。
| Origin绘制法
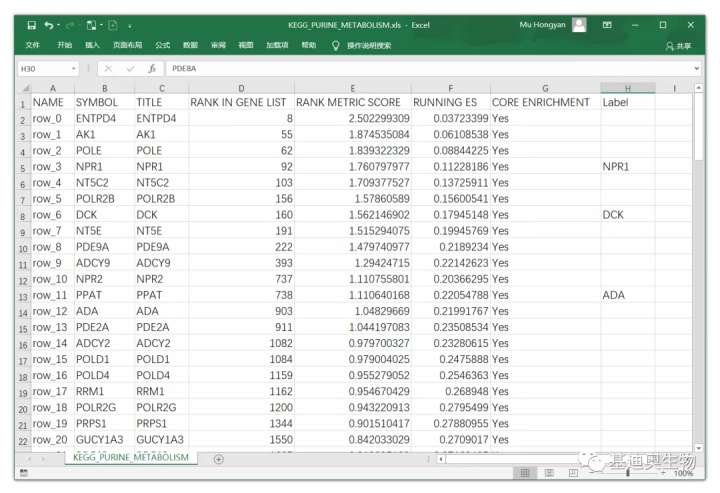
首先,用Excel打开GSEA分析结果中需要重绘的基因集作图数据,在原数据的基础上新增Label列,填入自己感兴趣的基因,如下图。
然后将范例数据复制粘贴到Origin(版本Origin 2021)中表格中,通过Plot/Basic 2D/Color Mapped绘制散点图,如下图。
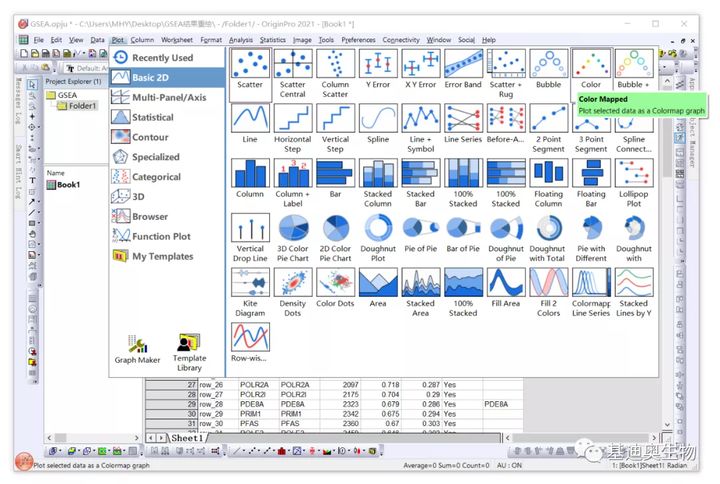
在Plot Setup窗口,给X轴、Y轴、映射颜色(C)、标签(L)选择绘图数据,方法如下,点OK按钮完成绘制。
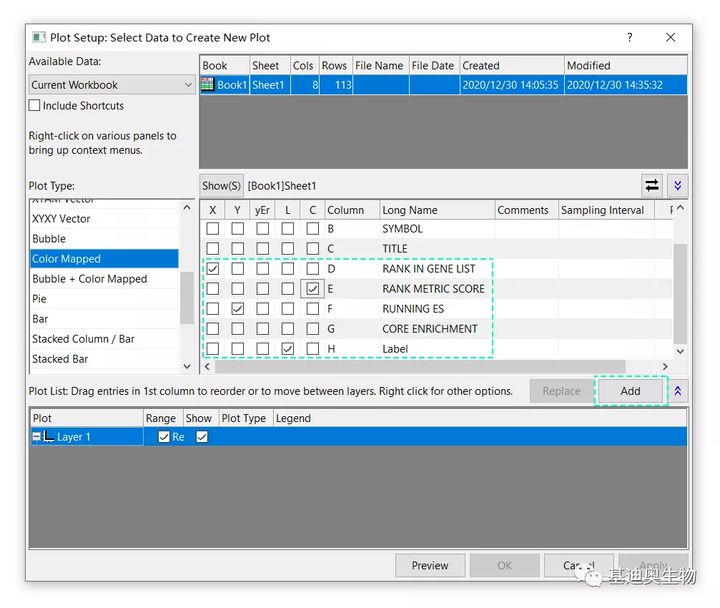
初始的结果如下:
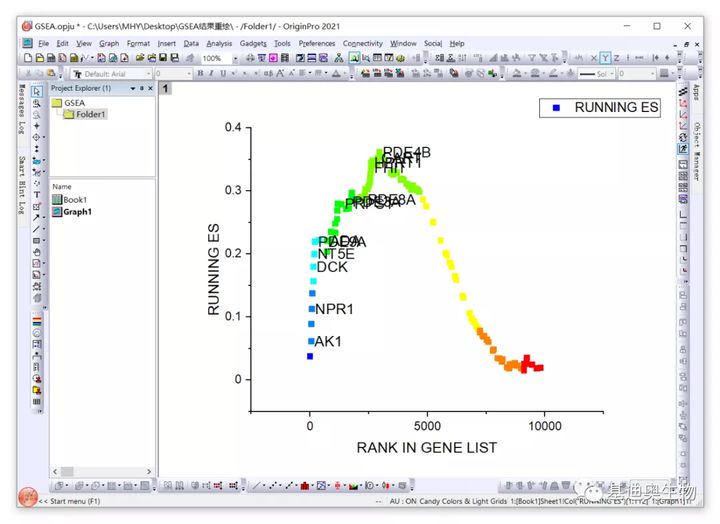
双击图表,在Plot Details窗口的Symbol选项下可调整散点的形状,大小(Size),Plot Type,如下。
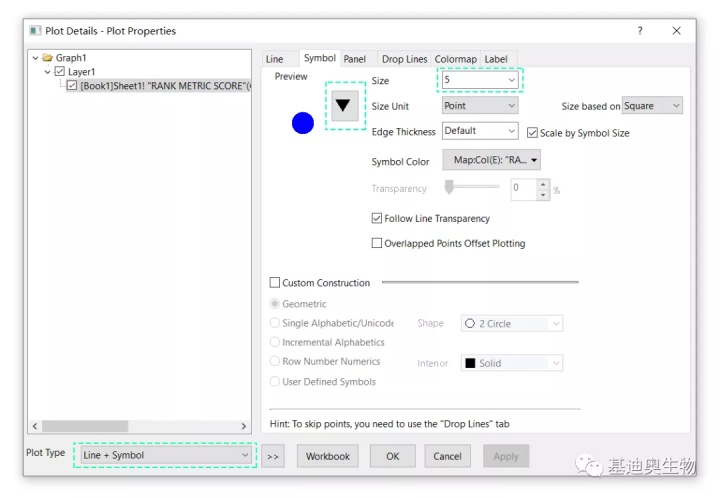
接续在Line选项下调整连线的粗细和渐变色,方法如下。
接着,在DropLines选项下勾选Vertical,并重新设置渐变颜色,如下。
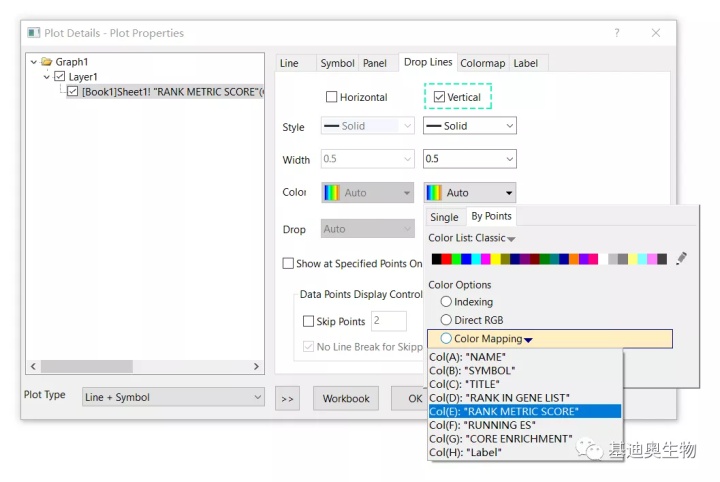
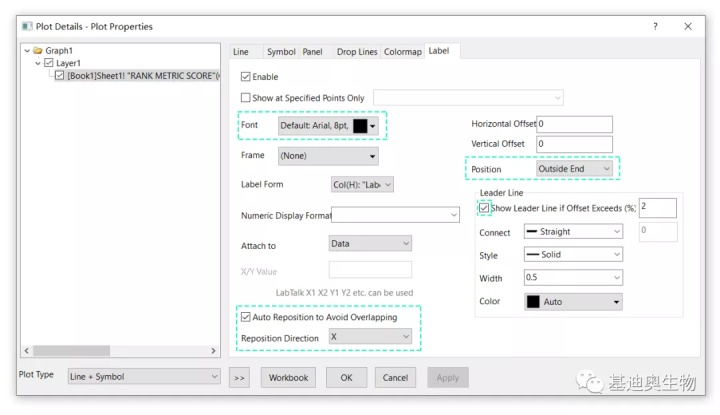
最后,在label选项下调整字体的大小,我这里设为8pt,同时设置标签的位置和指引线,勾选标签避免重叠,具体参数设置如下图。
调整后的效果如下:
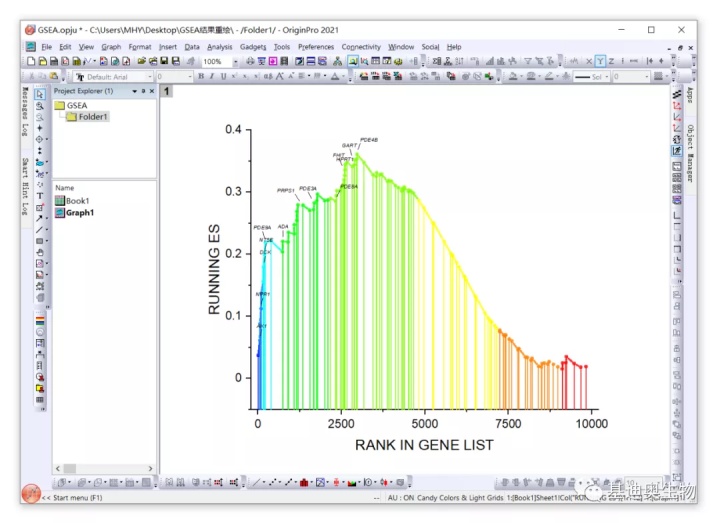
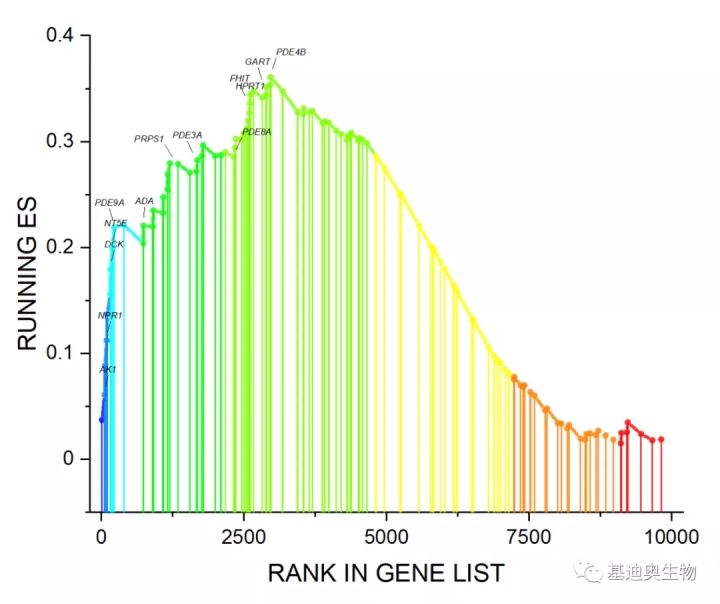
如果觉得颜色数量太多了,可双击图表,在Plot Details窗口调整配色,比如我这里设为“黄色-青色”渐变。
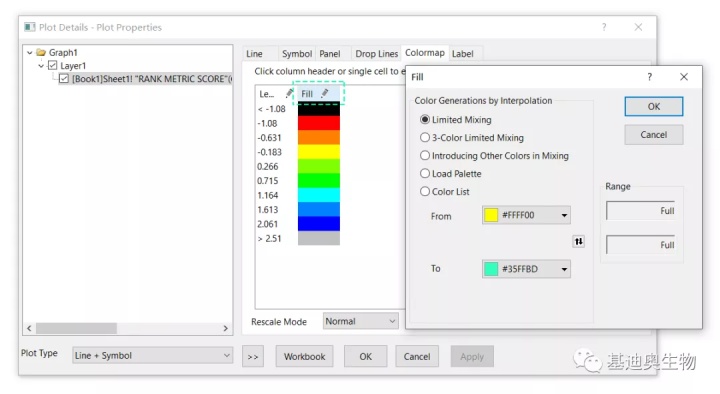
连续点击peak基因对应的散点3次,可单独对某个点进行调整,比如我这里将散点和drop line的颜色设置为橙色。

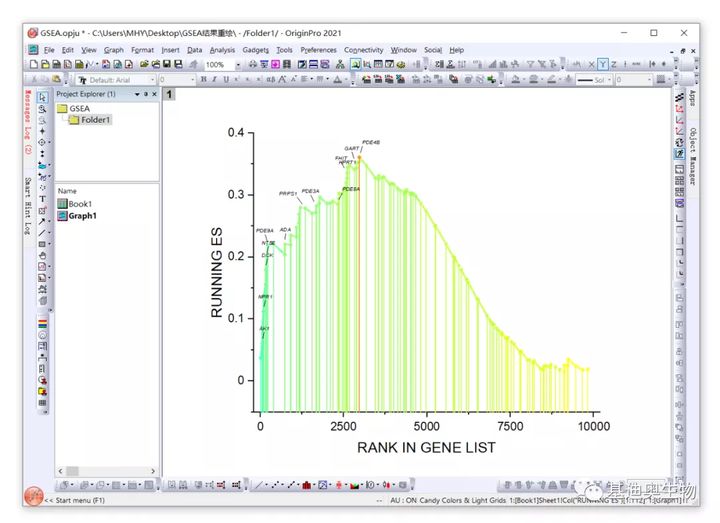
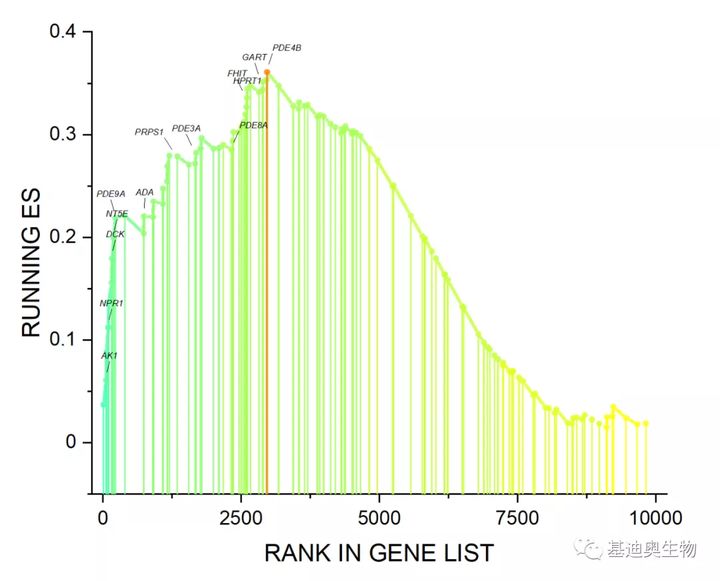
调整后的效果如下:
| ggplot2绘制法
关于绘制原理,主要是使用ggplot2包绘制折线图(geom_line+geom_point),然后再为每个散点添加竖线(geom_segment)。下面就先读入范例数据,载入ggplot2包,开始绘制吧。
#改变工作目录;
setwd("C:/Users/MHY/Desktop/GSEA结果重绘")
#读入GSEA结果数据;
df <- read.table("KEGG_PURINE_METABOLISM.xls",header = T,sep = 't')
head(df)
#载入ggplot2包;
library(ggplot2)
#建立映射,绘制图形区;
p<- ggplot(df, aes(x=RANK.IN.GENE.LIST,
y=RUNNING.ES,
colour=RANK.METRIC.SCORE))
p1 <- p + geom_line(size=0.5) + geom_point(size=0.5)
p1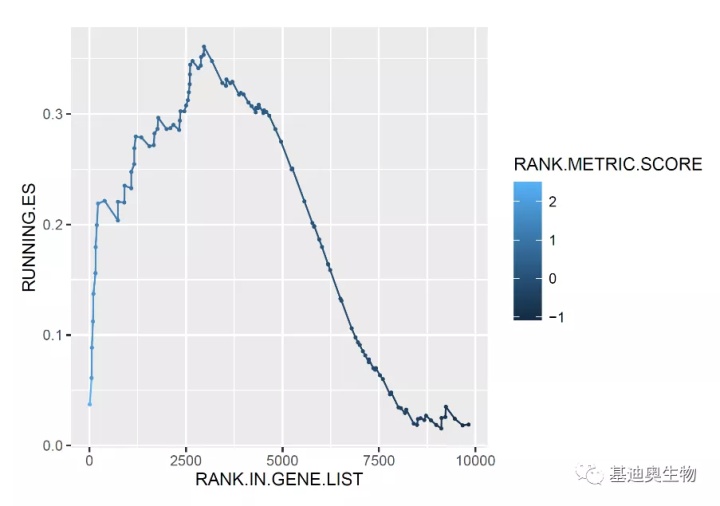
#添加竖线;
p2 <- p1 + geom_segment(data=df,
aes(xend=RANK.IN.GENE.LIST, yend=0), size=0.4)
p2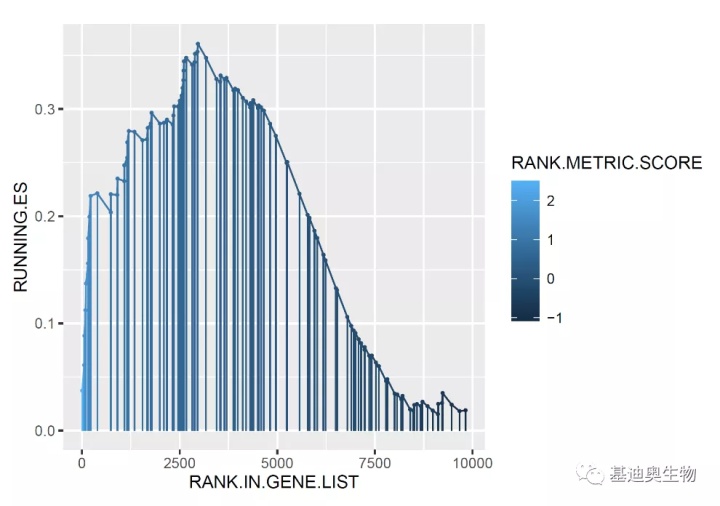
#使用2种渐变色;
p3 <- p2+scale_color_gradient(low = "cyan", high = "yellow",
guide=guide_colorbar(title="Rank",
title.position="top",
barwidth=0.7,
barheight = 5,
frame.colour = NULL,
ticks = T))
p3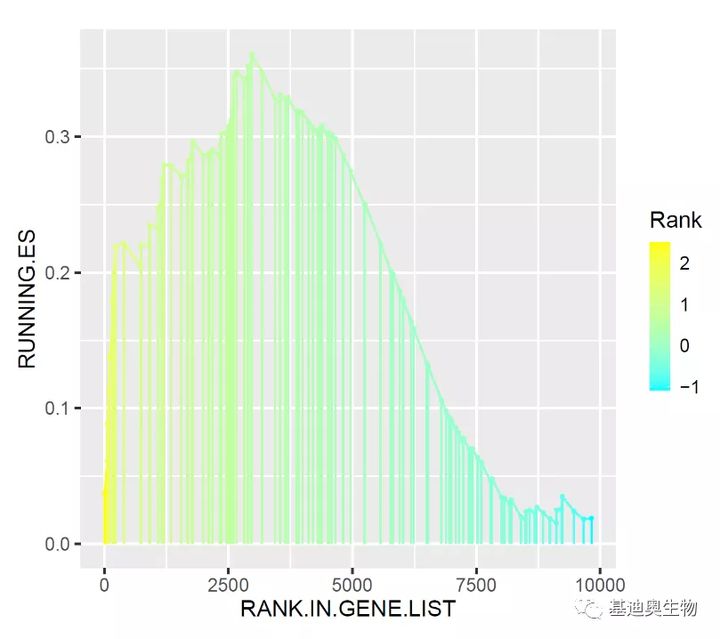
#使用多种渐变色;
p3 <- p2 + scale_color_gradientn(
colours = rainbow(6),
guide=guide_colorbar(title="Rank",
title.position="top",
barwidth=0.7,
barheight = 5,
frame.colour = NULL,
ticks = T))
p3
#提取核心基因集(leading edge genes);
df_genes <- subset(df,CORE.ENRICHMENT=="Yes")
head(df_genes)
#提取peak基因;
peak <- tail(df_genes,1)
#突出展示peak基因;
p4 <- p3 + geom_point(data=peak,
aes(x=RANK.IN.GENE.LIST,y=RUNNING.ES),
colour="tomato", alpha=1, size=1) +
geom_segment(data=peak,
aes(xend=RANK.IN.GENE.LIST, yend=0),
size=0.5, colour="tomato")
p4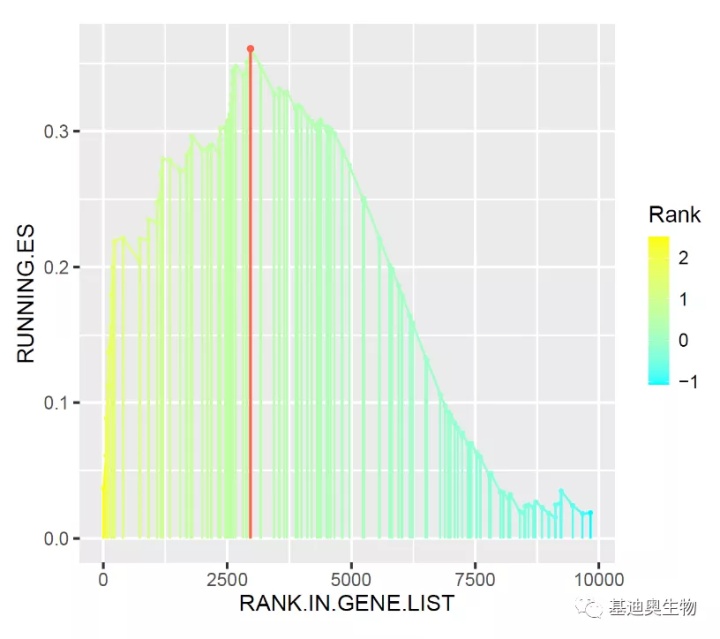
接下来使用ggrepel包为图表添加文字标签和指引线,关于这个包的详细用法可参考之前的文章:
基迪奥生物:标签糊成一坨?如何为图表添加“帅帅”的指引线?zhuanlan.zhihu.com
library(ggrepel)
#指引线的参数设置;
leader.size <- 2
leader.color <- "grey10"
leader.alpha <- 0.9
p5 <- p4 + geom_text_repel(data=df_genes,
aes(x=RANK.IN.GENE.LIST,y=RUNNING.ES,label=SYMBOL),
lineheight=0.8,
size=leader.size,
color=leader.color,
alpha=leader.alpha,
fontface='italic',
box.padding=unit(0.5,"lines"),
segment.color='grey30',
segment.alpha=0.5,
segment.size=0.5)
p5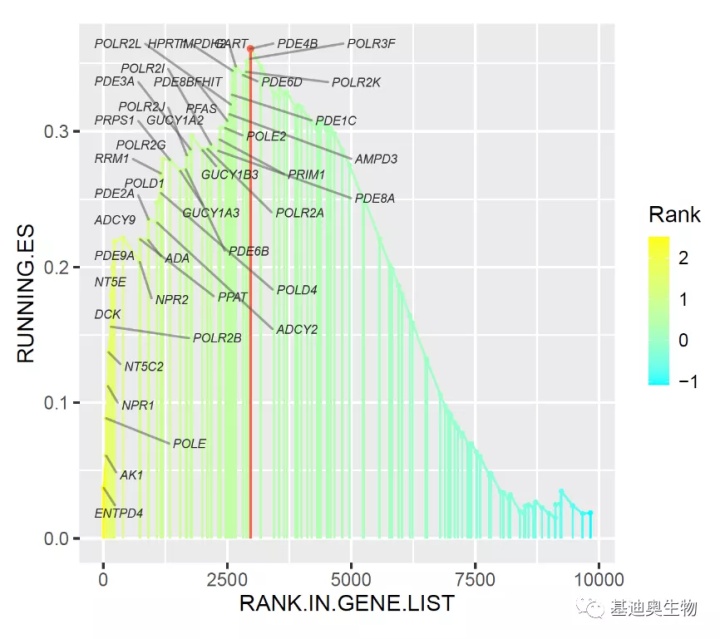
#添加带箭头的指引线;
p6 <- p4+geom_text_repel(data=df_genes,
aes(x=RANK.IN.GENE.LIST,y=RUNNING.ES,label=SYMBOL),
force=0.3,
color=leader.color,
size=leader.size,
fontface='italic',
point.padding = 0.5,
hjust = 0.5,
arrow = arrow(length = unit(0.01,"npc"), type ="open", ends ="last"),
segment.color=leader.color,
segment.size=0.2,
segment.alpha=leader.alpha,
nudge_y=0)
p6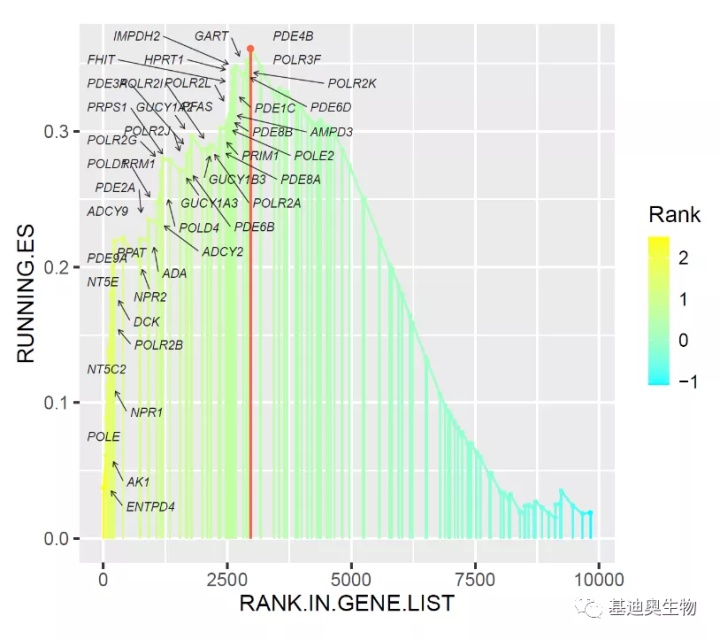
我们可以发现,大量的文字标签都堆积在图表的左半部分,即便使用ggrepel包,仍有部分标签重叠在一起,而且,整个图表我们最关心的是leading edge。如何突出展示这一区域呢?
你可以像范例文献那般,只绘制这一区域的基因,还可以对整个gene set的rank值取对数,对这一区域进行视觉“放大”。
#将x坐标轴转换为对数型坐标轴;
library(scales)
x <- .x <- NULL
#对坐标轴进行log10对数转化;
p7 <- p6 + scale_x_log10(breaks=trans_breaks("log10", function(x) 10^x, n=4), labels = trans_format("log10",math_format(10^.x)))+
scale_y_continuous(limits = c(0, 0.42),expand = c(0.001,0))
p7取对数后的作图效果如下:
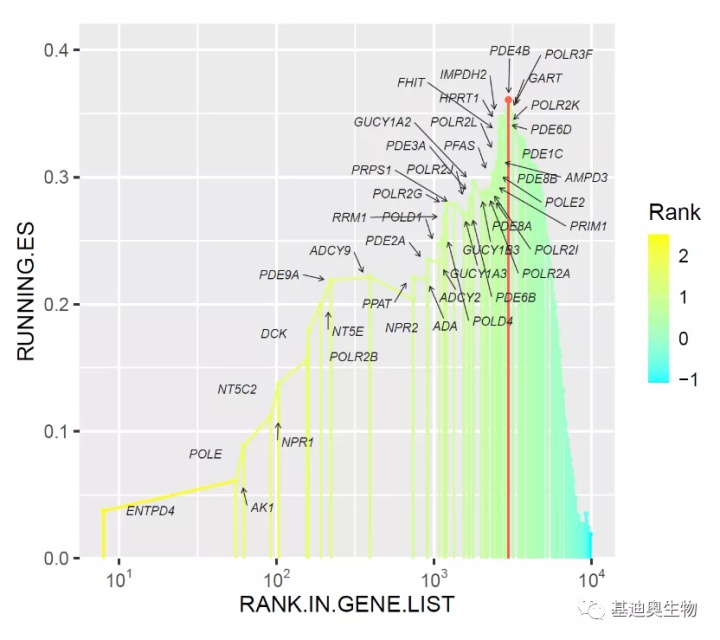
#修改主题样式;
p8 <- p7 + theme_classic() +
theme(axis.title=element_text(color="black"),
axis.text =element_text(color="black"),
legend.title = element_text(size = 8),
axis.line =element_line(size = 0.5))
pic <- p8+xlab("Gene ranks") + ylab("Running enrichment score")
pic最终的绘图效果如下:
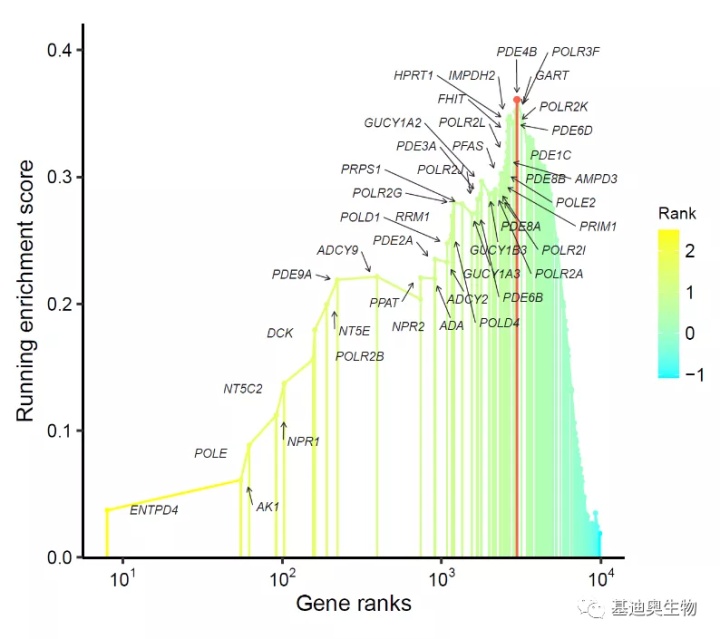
| 作图数据准备
本文的范例数据来自OmicShare GSEA工具的分析结果,具体获得过程请参考《高端的GSEA往往只需采用最简单的分析工具》一文。我这里仅从分析结果中随便选了一个基因集的作图数据(即KEGG_PURINE_METABOLISM.xls)进行重新绘制,如下图。
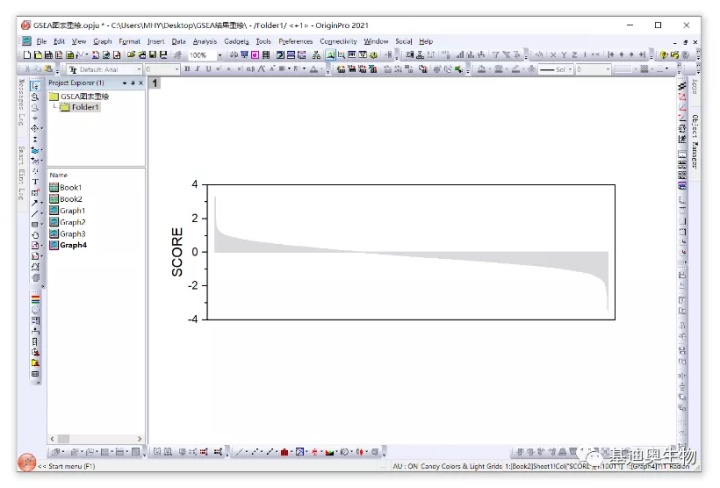
当然如果想绘制ES组合图下方的排序得分柱状图,可以使用的“ranked_gene_list.xls”文件进行绘制,如下。
具体可绘制出这样的柱状图:
所有组合图下半部分的柱状图都是相同的,因此,只绘1个即可。然后,与其他的基因集ES折线图进行组合,效果如下:
好啦,如果你还不会做基因集富集分析(GSEA),可试试OmicShare新推出的两款在线小工具,数据准备方法简单,除了人,还支持水稻、拟南芥等多种模式生物!
OmicShare Tools - 基迪奥生物信息绘图云平台www.omicshare.com
率先观看更多工具使用教程,欢迎关注官方微信公众号:基迪奥生物 (gene-denovo)
参考资料
Fairfax B P , Taylor C A , Watson R A , et al. Peripheral CD8+ T cell characteristics associated with durable responses to immune checkpoint blockade in patients with metastatic melanoma[J]. Nature medicine, 2020, 26(11).

















 1304
1304

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









