





无需写代码的高颜值富集分析神器
富集分析是了解一个基因集功能倾向性的一个方式,在组学研究领域应用广泛。常见的有基于差异基因的Over-representation分析,也就是常说的GO、KEGG富集分析和Functional class scoring分析,如GSEA。这两种富集分析算法不同,但可以都支持同样的注释集,如GO、KEGG或其他类型的注释。基本原则只有一个:基因集的基因名字与注释集的基因名字能匹配。剩下的就是了解下原理去操作了。
下面三篇文章建议仔细多读几遍,尤其是原理解释部分。对以后的分析事半功倍。做了一年的培训的体验是:学不会是因为没时间。时间用到了,自然就会了,培训现场的进度就是最好的支持证据。我们的培训希望在3天时间告诉大家需要3个月到半年可以掌握的知识,多读几遍也就都会了 (跑题了)。
- GO、GSEA富集分析一网打进
- GSEA富集分析 - 界面操作
- 去东方,最好用的在线GO富集分析工具
今天给大家介绍的是Cytoscape的插件??ClueGO。ClueGO能以网络图的形式来展示GO富集结果。
除了在线工具和R代码,今天带来的是网络绘制工具Cytoscape的一个插件ClueGO,支持200多个物种,支持数据库实时更新,支持多个注释数据集,支持多集合富集结果比较和网络图展示。配合Cytoscape的网络操作,可以做更多探索。下面看看怎么安装和使用吧。
ClueGO安装
官网下载
(http://apps.cytoscape.org/apps/cluego)
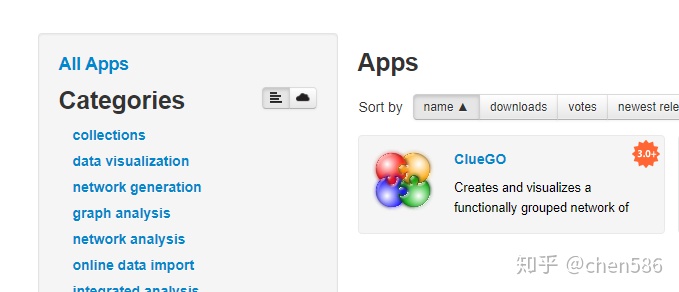
2.Cytoscape的APP Manager中下载
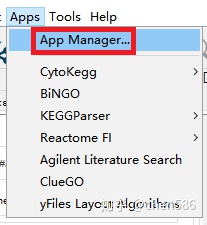
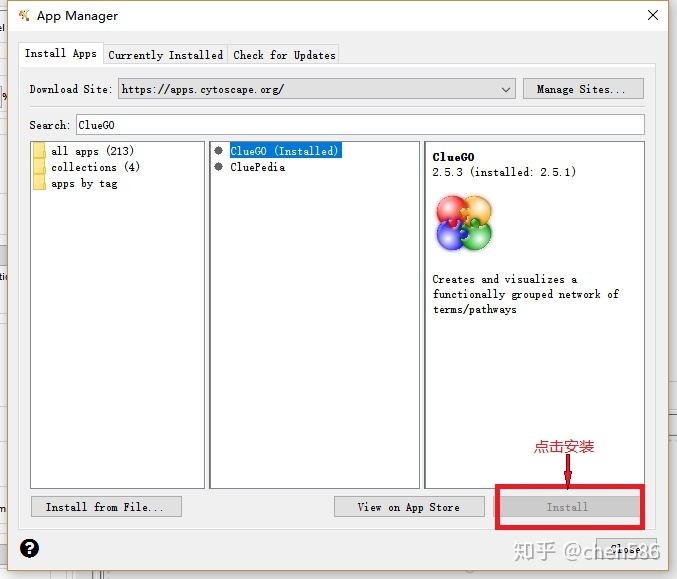
ClueGO激活
ClueGO第一次使用需要按照提示申请license,申请网址为:http://www.ici.upmc.fr/cluego/cluegoLicense.shtml。 点击Request a license key,填写基本信息之后提交审核,审核时间可能需要几天。拿到license之后点击App-ClueGO,在弹出的提示框中输入license即可激活ClueGO。
简单使用ClueGO
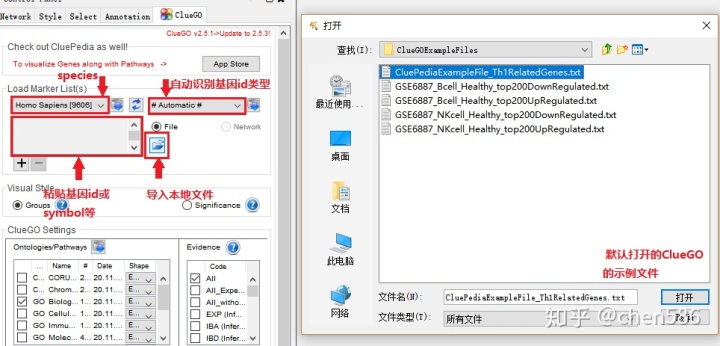
1. 上传数据
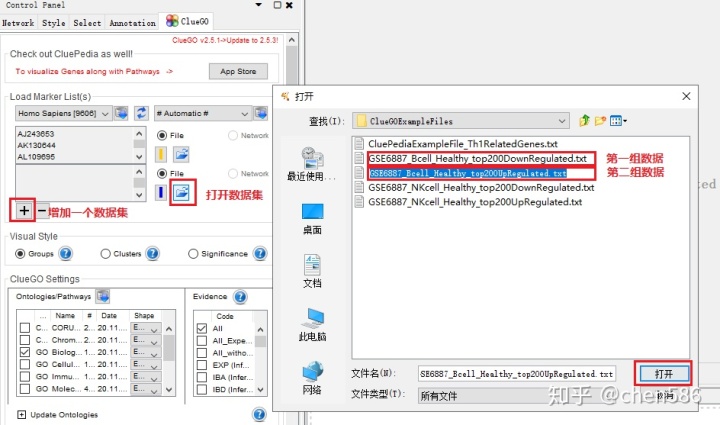
可以粘贴基因的id、symbol等或从本地上传文件 (富集分析的基因集只需要基因名字),我们以ClueGO的示例文件“CluePediaExampleFile_Th1RelatedGenes.txt”为例,并选择其对应物种和自动识别Gene id类型选项。
2. 选择富集数据集和富集标准
默认是BP,我们增加选择了CC和MF。同时可以在右侧选择不同支持度的注释,实验支持的、人工校对的还是计算预测的等。GO中的IEA数据为电子注释结果。如果物种注释的好,如模式生物,可以比较下有无IEA对富集结果的影响。
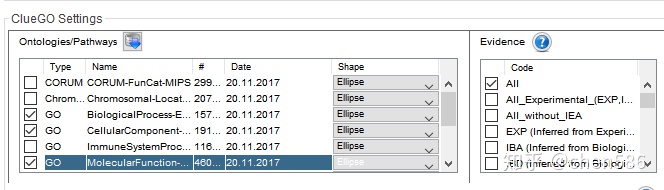
然后勾选以下选项,目的是只显示p值<=0.05的富集结果。具体选择的富集显著性Pvalue,可自行调整。

3. 富集分析和结果展示
点击start开始进行富集分析
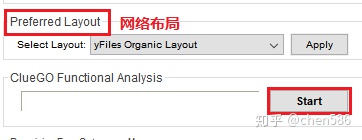
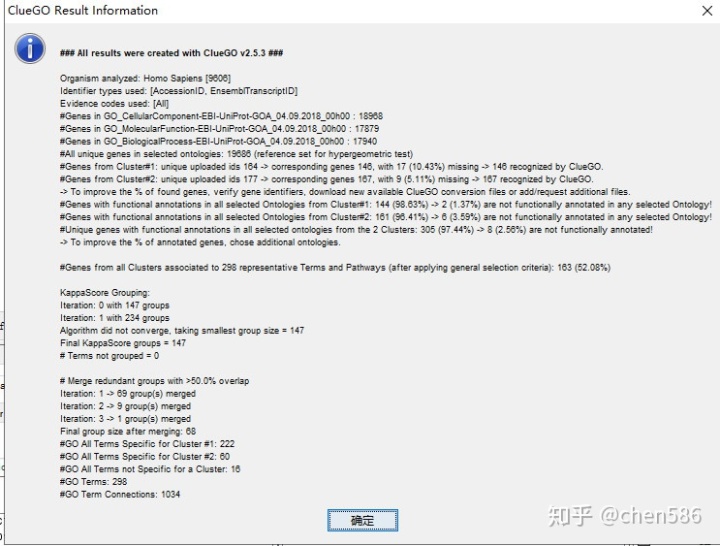
运行结束后,会跳出一个简述富集结果信息的提示框,包括物种, 基因名字,匹配的注释信息,重叠度和冗余度等信息。
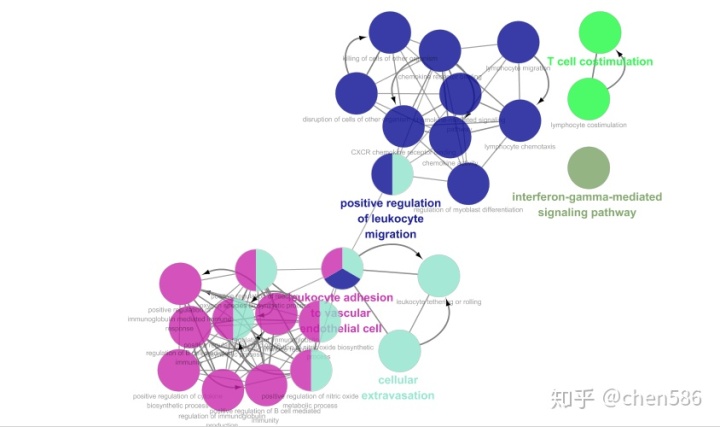
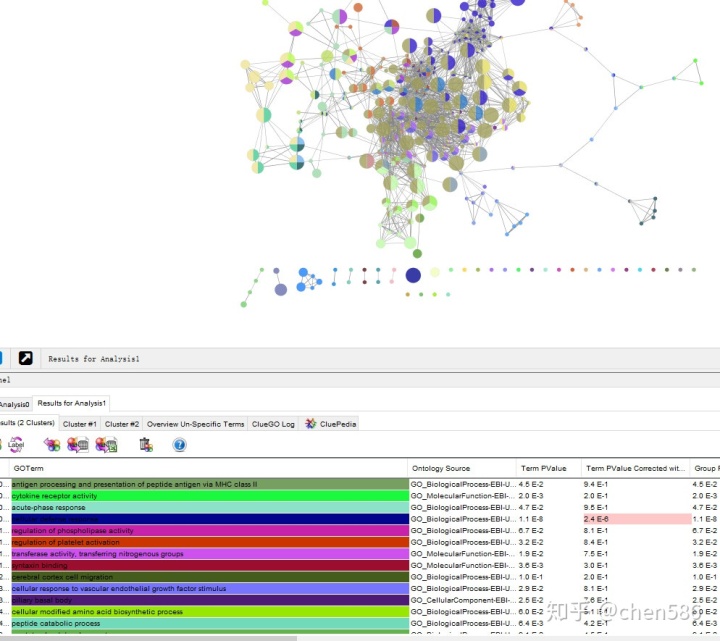
Cytoscape的网络视图中展示的是富集结果的网络图。每个节点为代表性富集通路,节点的连线代表通路之间的共有的基因数,颜色代表该节点的富集情况分类(隶属于哪个功能组,颜色和表格形式的的ClueGO结果对应)。
4. 结果导出矢量图和表格
结果导出为矢量图

结果保存为表格或Excel格式

ClueGO优势
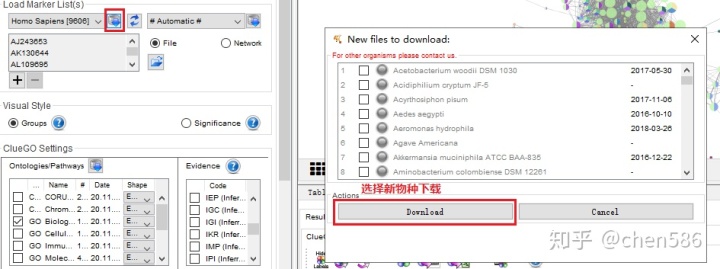
1. 物种丰富,并支持手动下载
本地有人类和小鼠可选,也可根据需要下载,支持近200个物种。
2. 物种注释手动更新
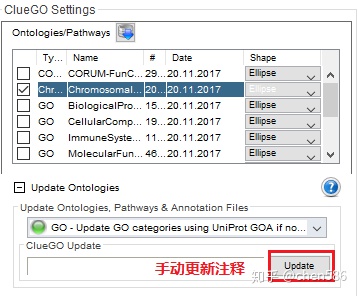
(前面推荐的GOEAST是每周更新,这个可以认为是实时更新。R中的注释包相比来说更新的慢了些,需要社区发布新版本或自己整理。)
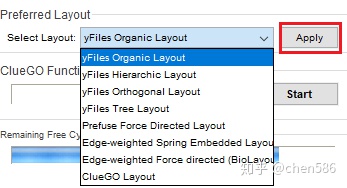
3. 多种网络布局和自由网络设计
网络布局类型选择
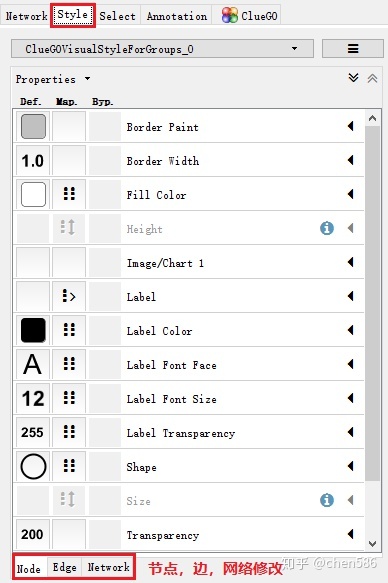
style选项可对网络的颜色,节点,网络等进行修改。还可以映射其它如表达量信息等。
4. 多个数据集比较
同时导入2组或多组基因列表,可以进行比较分析。
富集结果
信息概括
网络图和详细信息
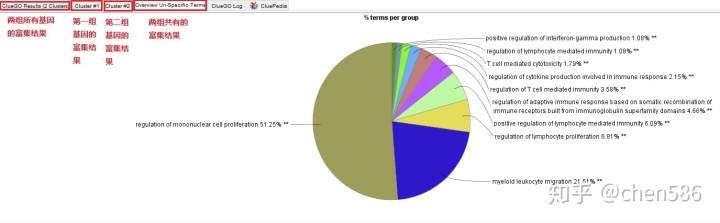
5. 链接外源数据库
看到感兴趣的基因、通路,可以直接选择需要的外源数据库查询获得更多信息。

更多Cytoscape的使用
- Cytoscape教程1
- Cytoscape之操作界面介绍
- 新出炉的Cytoscape视频教程
- Cytoscape制作带bar图和pie图节点的网络图
- Cytoscape: MCODE增强包的网络模块化分析
- 生信宝典之傻瓜式 (五) 文献挖掘查找指定基因调控网络















 3893
3893











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









