







续前文“R包circlize绘制特定SNP、InDel位点标识的基因组圈图续前文“
R包circlize绘制基因组重测序变异圈图示例mp.weixin.qq.com
”。在前文绘制基因组圈图时,由于SNP、InDel过多,没有选择将这些变异位点单独以点的形式标记在基因组的特定位置处,而是统计了每2000bp滑窗中SNP、InDel位点的密度并绘制了密度分布。如下所示,详情参考前文。
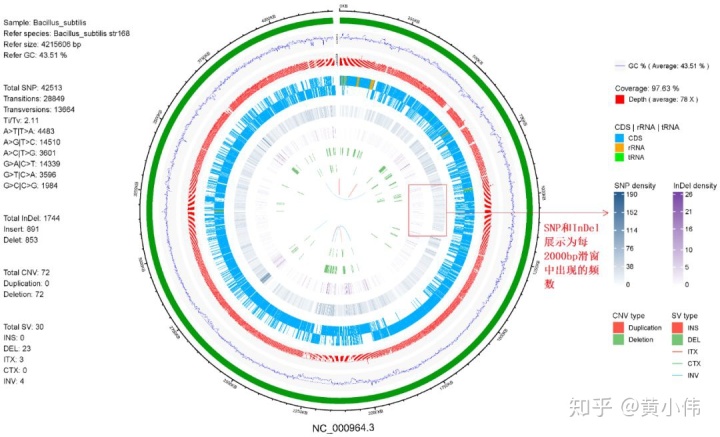
当时还提到,后续会再展示如何标注独立的SNP或InDel位点,而非密度频数。
然后就忘了……

上周有同学咨询,在提供帮助的同时就顺便简单整理了方法,在本篇继续分享。
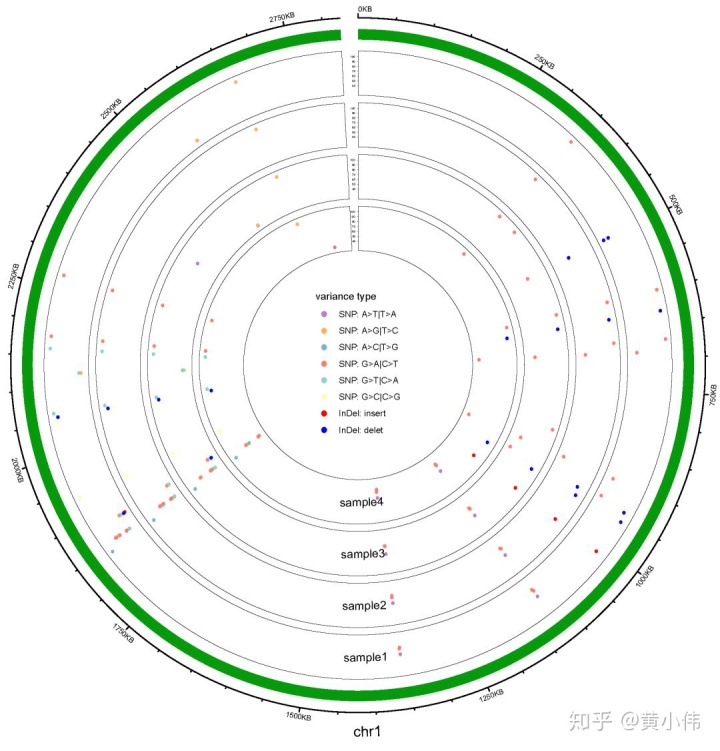
示例图大致如下。sample1-4是4个同种不同菌株细菌基因组测序样本,chr1是参考细菌基因组的染色体,并通过与参考基因组比对call变异,确定并筛选了一些SNP以及InDel位点。由于筛选后的位点不多,就方便了直接在基因组对应位置中将它们标注出来。
该图将4个菌株放在一起作图比较,每个圈代表各自基因组。图中的点即为SNP或InDel位点,颜色表示不用的变异类型(如SNP的A>C|T>G替换,或者InDel的插入缺失等)。点对应外圈中的刻度即为变异位点的基因组位置,纵轴数值表示了变异位点的质量。
接下来开始本篇的分享。
作图示例数据及R代码的百度盘链接(提取码,mgse):
https://pan.baidu.com/s/1_nKWA4I5QfUN9538hDqjsQ
示例数据
示例文件“genome_stat.txt”记录了参考基因组信息,即基因包含的染色体ID以及该染色体的长度。

示例文件“sample*_SNV.txt”的内容大致如下。

第1列,变异位点所在的染色体ID;
第2-3列,变异位点在该染色体中的起始和终止位置,SNP是单碱基,InDel可能是多碱基;
第4列,变异类型,SNP就是碱基替换类型,InDel就是插入缺失类型;
第5列,该位点中,变异碱基的占比,在本示例中用于绘制y刻度轴。
以及示例文件“anno_SNV.txt”,在上述基础上添加了一列注释信息,例如SNP导致了同义突变还是非同义突变等。

示例作图1,直接标记变异位点区块
首先先展示一种简单的绘制方法。并非绘制二维坐标点图,而是类似前文变异圈图的那种样式,通过色块展示位点。区别在于,前文的色块展示的变异位点出现频数,这里直接标记具体位置的位点。
首先预处理数据。
#读取数据
seq_stat <- read.delim('genome_stat.txt', stringsAsFactors = FALSE)
sample1 <- read.delim('sample1_SNV.txt', stringsAsFactors = FALSE)
sample2 <- read.delim('sample2_SNV.txt', stringsAsFactors = FALSE)
sample3 <- read.delim('sample3_SNV.txt', stringsAsFactors = FALSE)
sample4 <- read.delim('sample4_SNV.txt', stringsAsFactors = FALSE)
#统计变异类型
sample_df <- function(dat) {
dat[which(dat$change == 'A>T'|dat$change == 'T>A'),'type'] <- 1
dat[which(dat$change == 'A>G'|dat$change == 'T>C'),'type'] <- 2
dat[which(dat$change == 'A>C'|dat$change == 'T>G'),'type'] <- 3
dat[which(dat$change == 'G>A'|dat$change == 'C>T'),'type'] <- 4
dat[which(dat$change == 'G>T'|dat$change == 'C>A'),'type'] <- 5
dat[which(dat$change == 'G>C'|dat$change == 'C>G'),'type'] <- 6
dat[which(dat$change == 'insert'),'type'] <- 7
dat[which(dat$change == 'delet'),'type'] <- 8
return(dat[c(1:3, ncol(dat))])
}
sample1_df <- sample_df(sample1)
sample2_df <- sample_df(sample2)
sample3_df <- sample_df(sample3)
sample4_df <- sample_df(sample4)
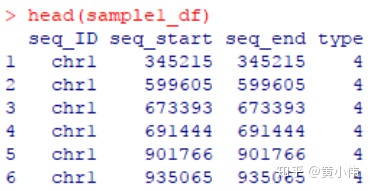
每张表格处理成4列,包含染色体ID,变异位点在该染色体中的起点和终点位置,以及变异类型。在本示例中,我将不同类型的SNP或InDel以不同数值指代。
然后绘制基因组圈图。
library(circlize) #圈图绘制
library(ComplexHeatmap) #这个包绘制图例
library(grid) #组合圈图和图例
pdf('circlize1.pdf', width = 8, height = 8)
circle_size = unit(1, 'snpc')
circos.par(gap.degree = 3, start.degree = 90)
#染色体区域
circos.genomicInitialize(seq_stat, plotType = c('axis', 'labels'), major.by = 250000, track.height = 0.05)
circos.genomicTrackPlotRegion(
seq_stat, track.height = 0.05, stack = TRUE, bg.border = NA,
panel.fun = function(region, value, ...) {
circos.genomicRect(region, value, col = '#049a0b', border = NA, ...)
} )
#sample1 SNV
color_assign <- colorRamp2(breaks = c(1, 2, 3, 4, 5, 6, 7, 8), color = c('#BC80BD', '#FDB462', '#80B1D3', '#FB8072', '#8DD3C7', '#FFFFB3', 'red', 'blue'))
circos.genomicTrackPlotRegion(
sample1_df, track.height = 0.1, stack = TRUE, bg.border = NA,
panel.fun = function(region, value, ...) {
circos.genomicRect(region, value, col = color_assign(value[[1]]), border = NA, ...)
xlim = CELL_META$xlim
ylim = CELL_META$ylim
circos.text(mean(xlim), mean(ylim), 'sample1', cex = 0.7, col = 'black', facing = 'inside', niceFacing = TRUE)
} )
#sample2 SNV
circos.genomicTrackPlotRegion(
sample2_df, track.height = 0.1, stack = TRUE, bg.border = NA,
panel.fun = function(region, value, ...) {
circos.genomicRect(region, value, col = color_assign(value[[1]]), border = NA, ...)
xlim = CELL_META$xlim
ylim = CELL_META$ylim
circos.text(mean(xlim), mean(ylim), 'sample2', cex = 0.7, col = 'black', facing = 'inside', niceFacing = TRUE)
} )
#sample3 SNV
circos.genomicTrackPlotRegion(
sample3_df, track.height = 0.1, stack = TRUE, bg.border = NA,
panel.fun = function(region, value, ...) {
circos.genomicRect(region, value, col = color_assign(value[[1]]), border = NA, ...)
xlim = CELL_META$xlim
ylim = CELL_META$ylim
circos.text(mean(xlim), mean(ylim), 'sample3', cex = 0.7, col = 'black', facing = 'inside', niceFacing = TRUE)
} )
#sample4 SNV
circos.genomicTrackPlotRegion(
sample4_df, track.height = 0.1, stack = TRUE, bg.border = NA,
panel.fun = function(region, value, ...) {
circos.genomicRect(region, value, col = color_assign(value[[1]]), border = NA, ...)
xlim = CELL_META$xlim
ylim = CELL_META$ylim
circos.text(mean(xlim), mean(ylim), 'sample4', cex = 0.7, col = 'black', facing = 'inside', niceFacing = TRUE)
} )
#图例
snv_legend <- Legend(
at = c(1, 2, 3, 4, 5, 6, 7, 8),
labels = c(' SNP: A>T|T>A', ' SNP: A>G|T>C', ' SNP: A>C|T>G', ' SNP: G>A|C>T', ' SNP: G>T|C>A', ' SNP: G>C|C>G', ' InDel: insert', ' InDel: delet'),
labels_gp = gpar(fontsize = 6), title = 'variance type', title_gp = gpar(fontsize = 7),
grid_height = unit(0.4, 'cm'), grid_width = unit(0.4, 'cm'), type = 'points', pch = NA,
background = c('#BC80BD', '#FDB462', '#80B1D3', '#FB8072', '#8DD3C7', '#FFFFB3', 'red', 'blue') )
pushViewport(viewport(x = 0.5, y = 0.5))
grid.draw(snv_legend)
upViewport()
circos.clear()
dev.off()

图中由外而内,依次是参考基因组范围,以及sample1-4中的变异位点,位点按类型上色。
通过比较,4种菌株中共有/特有变异一目了然……好吧看起来是有点费劲啊,这种绘制方法虽然简单,但是缺点还是蛮明显的。毕竟SNP只是特定位置的单碱基,InDel也只是少数连续的碱基插入或缺失,因此色块将非常难以辨认(输出的pdf矢量图,必须放大才能看到细节)。
示例作图2,通过散点标记变异位点
然后就是开篇展示的示例样式,以散点表示,将点尺寸设置的大一些就清楚了。
首先预处理数据,和上述标记为区块的方法是有区别的,复杂一些。
#读取数据
seq_stat <- read.delim('genome_stat.txt', stringsAsFactors = FALSE)
sample1 <- read.delim('sample1_SNV.txt', stringsAsFactors = FALSE)
sample2 <- read.delim('sample2_SNV.txt', stringsAsFactors = FALSE)
sample3 <- read.delim('sample3_SNV.txt', stringsAsFactors = FALSE)
sample4 <- read.delim('sample4_SNV.txt', stringsAsFactors = FALSE)
#统计变异类型
sample_list <- function(dat) {
dat <- list(dat[which(dat$change == 'A>T'|dat$change == 'T>A'), ],
dat[which(dat$change == 'A>G'|dat$change == 'T>C'), ],
dat[which(dat$change == 'A>C'|dat$change == 'T>G'), ],
dat[which(dat$change == 'G>A'|dat$change == 'C>T'), ],
dat[which(dat$change == 'G>T'|dat$change == 'C>A'), ],
dat[which(dat$change == 'G>C'|dat$change == 'C>G'), ],
dat[which(dat$change == 'insert'), ],
dat[which(dat$change == 'delet'), ])
return(dat)
}
sample1_list <- sample_list(sample1)
sample2_list <- sample_list(sample2)
sample3_list <- sample_list(sample3)
sample4_list <- sample_list(sample4)

这种方法中,由于需要按类型对点上色,同时还要保证y轴代表位点质量信息,按circlize的数据格式要求,需要整理为列表(list)样式。
本示例中,对于每个样本数据,按变异类型拆分为不同的数据框(data frame),每个示例中各自包含8种变异类型(6种碱基替换模式和2种插入缺失)因此各获得8个数据框,然后组合为列表。
然后绘制基因组圈图。
library(circlize) #圈图绘制
library(ComplexHeatmap) #这个包绘制图例
library(grid) #组合圈图和图例
pdf('circlize2.pdf', width = 8, height = 8)
circle_size = unit(1, 'snpc')
circos.par(gap.degree = 3, start.degree = 90)
#染色体区域
circos.genomicInitialize(seq_stat, plotType = c('axis', 'labels'), major.by = 250000, track.height = 0.05)
circos.genomicTrackPlotRegion(
seq_stat, track.height = 0.05, stack = TRUE, bg.border = NA,
panel.fun = function(region, value, ...) {
circos.genomicRect(region, value, col = '#049a0b', border = NA, ...)
} )
#sample1 SNV
color_assign <- c('#BC80BD', '#FDB462', '#80B1D3', '#FB8072', '#8DD3C7', '#FFFFB3', 'red', 'blue')
circos.genomicTrackPlotRegion(
sample1_list, track.height = 0.12, bg.border = 'black', bg.lwd = 0.4,
panel.fun = function(region, value, ...) {
circos.genomicPoints(region, value, pch = 16, cex = 0.5, col = color_assign[getI(...)], ...)
circos.yaxis(labels.cex = 0.2, lwd = 0.1, tick.length = convert_x(0.15, 'mm'))
xlim = CELL_META$xlim
ylim = CELL_META$ylim
circos.text(mean(xlim), mean(ylim), 'sample1', cex = 0.7, col = 'black', facing = 'inside', niceFacing = TRUE)
} )
#sample2 SNV
circos.genomicTrackPlotRegion(
sample2_list, track.height = 0.12, bg.border = 'black', bg.lwd = 0.4,
panel.fun = function(region, value, ...) {
circos.genomicPoints(region, value, pch = 16, cex = 0.5, col = color_assign[getI(...)], ...)
circos.yaxis(labels.cex = 0.2, lwd = 0.1, tick.length = convert_x(0.15, 'mm'))
xlim = CELL_META$xlim
ylim = CELL_META$ylim
circos.text(mean(xlim), mean(ylim), 'sample2', cex = 0.7, col = 'black', facing = 'inside', niceFacing = TRUE)
} )
#sample3 SNV
circos.genomicTrackPlotRegion(
sample3_list, track.height = 0.12, bg.border = 'black', bg.lwd = 0.4,
panel.fun = function(region, value, ...) {
circos.genomicPoints(region, value, pch = 16, cex = 0.5, col = color_assign[getI(...)], ...)
circos.yaxis(labels.cex = 0.2, lwd = 0.1, tick.length = convert_x(0.15, 'mm'))
xlim = CELL_META$xlim
ylim = CELL_META$ylim
circos.text(mean(xlim), mean(ylim), 'sample3', cex = 0.7, col = 'black', facing = 'inside', niceFacing = TRUE)
} )
#sample4 SNV
circos.genomicTrackPlotRegion(
sample4_list, track.height = 0.12, bg.border = 'black', bg.lwd = 0.4,
panel.fun = function(region, value, ...) {
circos.genomicPoints(region, value, pch = 16, cex = 0.5, col = color_assign[getI(...)], ...)
circos.yaxis(labels.cex = 0.2, lwd = 0.1, tick.length = convert_x(0.15, 'mm'))
xlim = CELL_META$xlim
ylim = CELL_META$ylim
circos.text(mean(xlim), mean(ylim), 'sample4', cex = 0.7, col = 'black', facing = 'inside', niceFacing = TRUE)
} )
#图例
snv_legend <- Legend(
at = c(1, 2, 3, 4, 5, 6, 7, 8),
labels = c(' SNP: A>T|T>A', ' SNP: A>G|T>C', ' SNP: A>C|T>G', ' SNP: G>A|C>T', ' SNP: G>T|C>A', ' SNP: G>C|C>G', ' InDel: insert', ' InDel: delet'),
labels_gp = gpar(fontsize = 6), title = 'variance type', title_gp = gpar(fontsize = 7),
grid_height = unit(0.4, 'cm'), grid_width = unit(0.4, 'cm'), type = 'points', background = NA,
legend_gp = gpar(col = c('#BC80BD', '#FDB462', '#80B1D3', '#FB8072', '#8DD3C7', '#FFFFB3', 'red', 'blue')) )
pushViewport(viewport(x = 0.5, y = 0.5))
grid.draw(snv_legend)
upViewport()
circos.clear()
dev.off()
图中由外而内,依次是参考基因组范围,以及sample1-4中的变异位点,位点按类型上色,同时还展示了位点质量信息。
通过比较,4种菌株中共有/特有变异及可信度等一目了然。
示例作图3,在2基础上延伸,通过点形状标识变异注释
接下来在示例图2的基础上进一步延伸。
上述已经通过颜色标识了SNP或InDel类型。
然后期望再将变异位点的注释信息添加进来,比方说SNP导致了同义突变或非同义突变,通过更改点的形状作进一步标识。
参考以下示例。
##预处理
#读取数据
seq_stat <- read.delim('genome_stat.txt', stringsAsFactors = FALSE)
anno <- read.delim('anno_SNV.txt', stringsAsFactors = FALSE)
#统计变异类型
change <- c('A>T|T>A', 'A>G|T>C', 'A>C|T>G', 'G>A|C>T', 'G>T|C>A', 'G>C|C>G')
type <- c('synonymous SNV', 'nonsynonymous SNV')
anno_list <- list()
length(anno_list) <- length(change) * length(type)
l = 0
for (i in change) {
for (j in type) {
l = l + 1
anno_list[[l]] <- anno[which(anno$change == i & anno$type == j), ]
}
}
##作图
library(circlize) #圈图绘制
library(ComplexHeatmap) #这个包绘制图例
library(grid) #组合圈图和图例
pdf('circlize3.pdf', width = 6, height = 6)
circle_size = unit(1, 'snpc')
circos.par(gap.degree = 3, start.degree = 90)
#染色体区域
circos.genomicInitialize(seq_stat, plotType = c('axis', 'labels'), major.by = 250000, track.height = 0.05)
circos.genomicTrackPlotRegion(
seq_stat, track.height = 0.05, stack = TRUE, bg.border = NA,
panel.fun = function(region, value, ...) {
circos.genomicRect(region, value, col = '#049a0b', border = NA, ...)
} )
#SNP 位点
color_assign <- c('#BC80BD', '#FDB462', '#80B1D3', '#FB8072', '#8DD3C7', '#FFFFB3')
pch_assign <- c(16, 1)
circos.genomicTrackPlotRegion(
anno_list, track.height = 0.2, bg.border = 'black', bg.lwd = 0.4,
panel.fun = function(region, value, ...) {
n = getI(...)
circos.genomicPoints(region, value, pch = pch_assign[ifelse(n%%2, n%%2, 2)], cex = 0.5, col = color_assign[ifelse(n%%6, n%%6, 6)], ...)
circos.yaxis(labels.cex = 0.2, lwd = 0.1, tick.length = convert_x(0.15, 'mm'))
xlim = CELL_META$xlim
ylim = CELL_META$ylim
circos.text(mean(xlim), mean(ylim), 'sample', cex = 0.7, col = 'black', facing = 'inside', niceFacing = TRUE)
} )
#图例
snv_legend <- Legend(
at = c(1, 2, 3, 4, 5, 6, 7, 8),
labels = c(' SNP: A>T|T>A', ' SNP: A>G|T>C', ' SNP: A>C|T>G', ' SNP: G>A|C>T', ' SNP: G>T|C>A', ' SNP: G>C|C>G', 'synonymous SNV', 'nonsynonymous SNV'),
labels_gp = gpar(fontsize = 5), title = 'variance typen', title_gp = gpar(fontsize = 6),
grid_height = unit(0.4, 'cm'), grid_width = unit(0.4, 'cm'), type = 'points', background = NA,
legend_gp = gpar(col = c('#BC80BD', '#FDB462', '#80B1D3', '#FB8072', '#8DD3C7', '#FFFFB3', 'gray', 'gray')),
pch = c(20, 20, 20, 20, 20, 20, 16, 1) )
pushViewport(viewport(x = 0.5, y = 0.5))
grid.draw(snv_legend)
upViewport()
circos.clear()
dev.off()

由于实际操作起来确实有点麻烦……因此该示例只简单地展示了一圈。
该示例数据中只包含SNP,且只包含同义突变和非同义突变两种注释,简化过程便于大家了解这种作图方法。图中SNP位点按碱基替换类型上色,实心点为同义突变,空心点为非同义突变,纵轴展示了位点质量信息。















 445
445











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









