






circlize绘制圈图
今天要说的是一个比较常用的图形,也就是圈图。最开始接触圈图是时候我就感觉圈图是极坐标化的条形图,但是在用ggplot绘图时总觉得调整的不太好看,所以就用别人现成的R包绘制就好了。

1.绘图前参数设置
rm(list = ls())
options(stringsAsFactors = F)
#install.packages('circlize')
library(circlize)
#清除circos绘图
circos.clear()
#参数
circos.par("track.height"=0.1,gap.degree=3, start.degree =90,
clock.wise = T,track.margin=c(0,0.02), cell.padding=c(0,0,0,0))
start.degree 绘图起始位置,90表示12点钟
gap.degree track左右间隔大小,gap.after等价
track.margin track上下间隔大小 c(,)
track.height 每个track的默认高度,和track.margin单位一样,都是半径的百分比
cell.padding 看不懂,默认吧 c(,)
clock.wise 排列方式,顺时针 T/F
xaxis.clock.wise x轴,顺时针 T/F
然后介绍以下自带的数据,包括mm10和hg19的
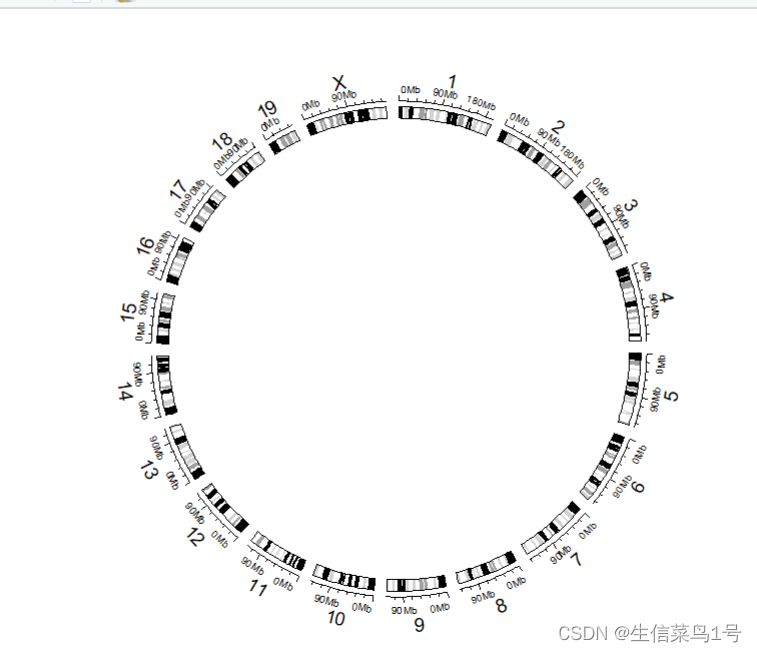
circos.initializeWithIdeogram(species = "mm10", #物种mm10,hg19
chromosome.index=c(paste0('chr',1:19),'chrX')
#选择染色体,默认全部,我们只用到1-19和x
)
然后就是绘图所需的数据了,包里有自带的示例数据,可以看到顺序就是chr start end value
bed = generateRandomBed(nr = 200)
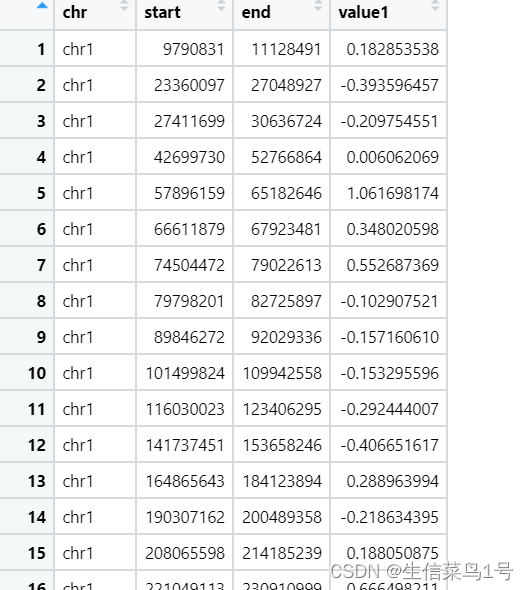
我们在准备数据的时候就按照这个来准备,绘制不同图形用不同数据,比如绘制线图点图有chr start value就好,如果绘制heatmap或者genomicRect这样的范围图则chr start end value都需要。
1.绘图
1.1 function(x,y)类型的绘图
function(x,y)类型的绘图包括折线图,点图等
1.1.1 折线图
方法1:在track中加入circos.lines绘制折线图
#绘图就用circos.track,这个函数表示新建一个层,需要参数染色体,x,y。里边circos.lines用于在层中绘制折线图。
circos.track(factors=bed$chr, x = bed$start, y = bed$value1,track.index=3,
track.height = 0.05,ylim=c(0,1),
panel.fun = function(x, y) {
circos.lines(x, y, col = "red") #默认折线图,x和y来自上边的参数传递
})
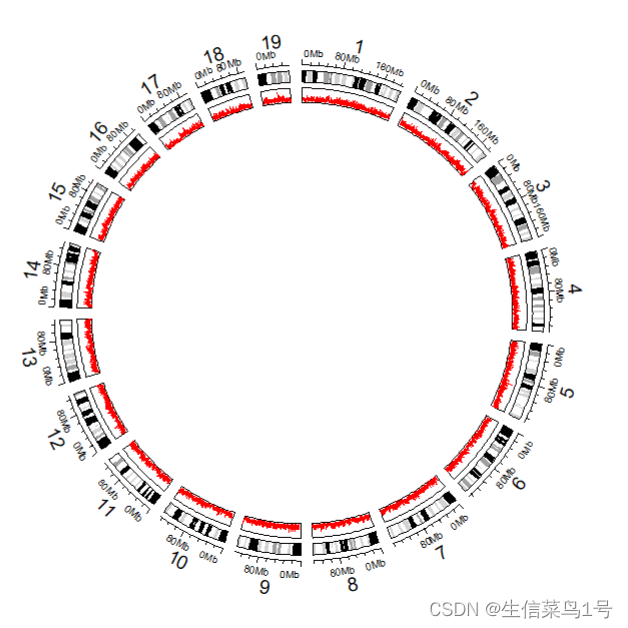
具体track里边的参数如下:
#注意Track的track.height很重要,且每个track的ylim需要设置
factors 染色体列
x 一般start列
y 一般value列,一般只有折线图点图之类的需要xy,其他都是具体区域
#factors,x和y有时候不需要设置,因为有的绘图需要的是region和value的格式
#只需要格式对了就好"chr",“start”,“end”,“value1”
ylim 限制y范围
track.index track索引,定义绘制在哪个track里
track.height 高度,在circos.par可以定义默认的,也可以在track里自定义
track.margin only affect current track
cell.padding only affect current track
bg.col = NA, track的背景色,可以是一个也可以是染色体数目个
bg.border = “black” track的边框,可以是NA
bg.lty = linetype
bg.lwd =linewidth
panel.fun 绘图函数 function(x,y) function(region,value)
circos.track函数相当于进行一个For循环,每次绘制一条染色体,所以在function内部可以用CELL_META s e c t o r . i n d e x 获取每次绘制时当前染色体的 i n d e x ,用 C E L L M E T A sector.index 获取每次绘制时当前染色体的index,用CELL_META sector.index获取每次绘制时当前染色体的index,用CELLMETAylim 获取y的范围,利用用CELL_META$xlim 获取x的范围,利用这一特点我们还可以为折线图绘制阈值线。
th=quantile(bed$value1,0.99)
circos.track(factors=bed$chr, x = bed$start, y = bed$value1,#track.index=3,
#bg.col=rainbow(19),
track.height = 0.05,ylim=c(0,1),
panel.fun = function(x, y) {
circos.lines(x, y, col = "red") #默认折线图
circos.segments(x0=CELL_META$xlim[1],x1=CELL_META$xlim[2], #添加segment
y0=th,y1=th,col = 'black')
})
})
添加阈值线如下,我这个没有定义track的index,不小心多运行了一次,如果不定义track的index绘制会依次绘制。

方法2:
先绘制一个空的track,再用circos.trackLines绘制折线图
circos.track(ylim=c(0,1),track.height = 0.05)
circos.trackLines(factors =bed$chr,x=bed$start,y=bed$value1,col='red')
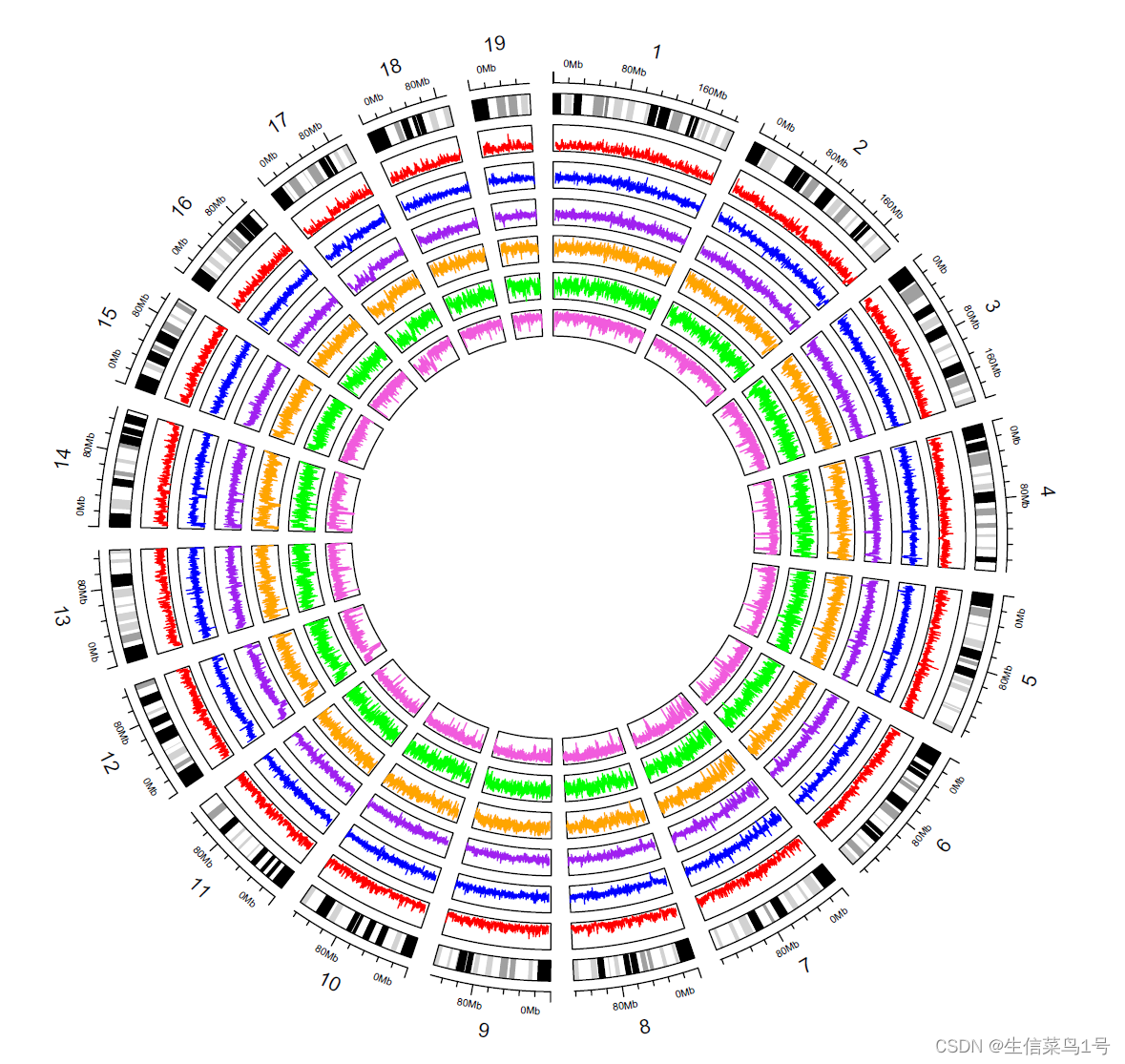
按照上面的函数多绘制几个track,每个用不同颜色的线表示,大概结果就如下图所示
1.1.2 点图
circos.track(factors=bed$chr, x = bed$start, y = bed$value1,#track.index=3,
#bg.col=rainbow(19),
track.height = 0.05,ylim=c(0,1),
panel.fun = function(x, y) {
circos.points(x, y, col = "red",cex =0.5)
circos.segments(x0=CELL_META$xlim[1],x1=CELL_META$xlim[2],
y0=th,y1=th,col = 'black')
})
自行调整点的大小
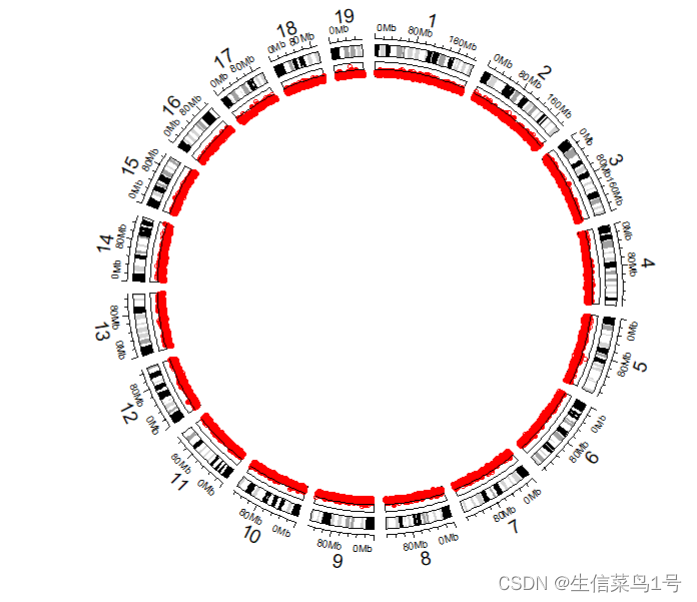
1.2 function(region,value)类型的绘图
这种绘图需要的数据是chr start end value,是定义某个范围内的数值大小
1.circos.genomicRect
circos.genomicTrack(bed,ylim=c(0,1), #bed是数据名,不用设置chr,x,y了
panel.fun = function(region, value, ...) { #resion就是 chr start end value就是value,排列顺序对了就ok
circos.genomicRect(region, value, ytop= 0.95, ybottom = 0.05,
#ytop.column 使用第几列value,或者自定义ytop
col = 'transparent',border='#2686e3',lwd=0.4)
#circos.lines(CELL_META$cell.xlim, c(0, 0), lty = 2, col = "#00000040")
}, stack = F,track.height = 0.05)
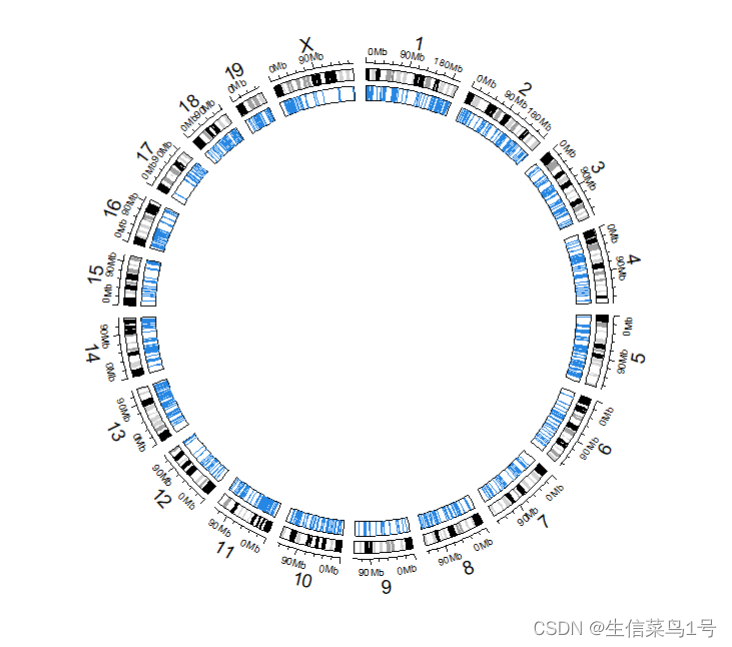
2.circos.genomicHeatmap
绘制某一区域的热图
col_fun = colorRamp2(c(0,1), c("green", "red"))
chr=c(paste0('chr',1:19),'chrX')
col=rainbow(20)
circos.genomicHeatmap(bed1, col = col_fun, side = "inside",
border = "white",
connection_height = mm_h(2),heatmap_height = 0.05,
line_col = col[match(bed1$chr,chr)]) #不同染色体用不同颜色绘制
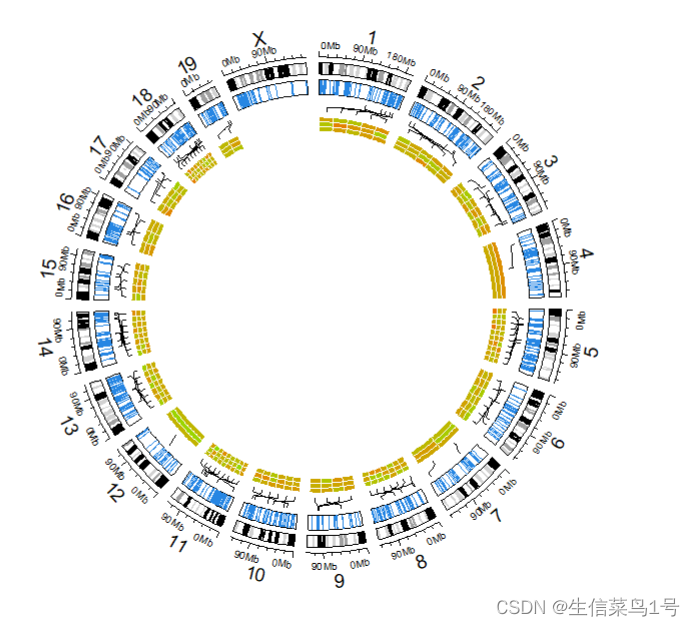
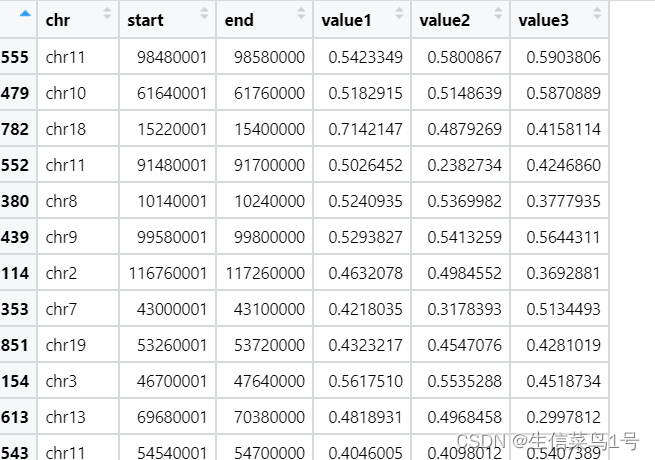
用到的数据bed1也是多个value数据
1.3 其他类型的绘图
1.直方图
大概的意思就是看一下你的数据分布情况
bgcol <- rep(c("#fb8072", "#80b1d3"), 10)
circos.trackHist(bed$chr,bed$start, bin.size = 1e7,
bg.col = bgcol, col = NA,track.height = 0.05)
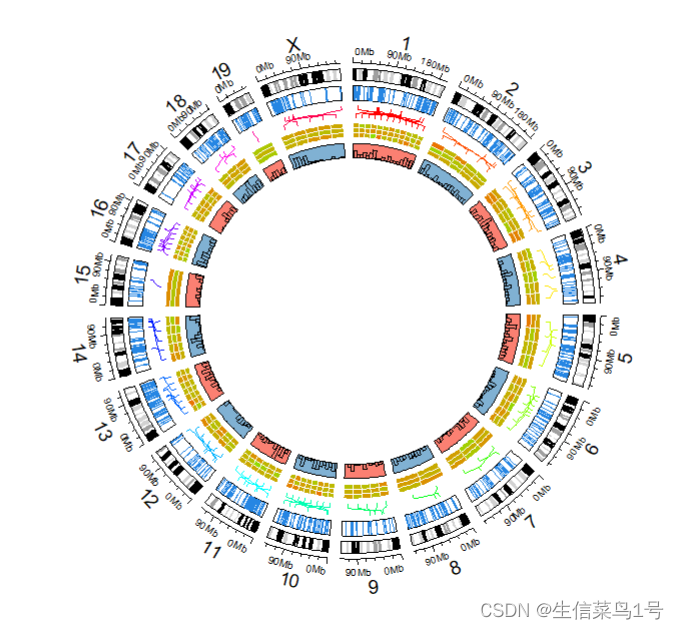
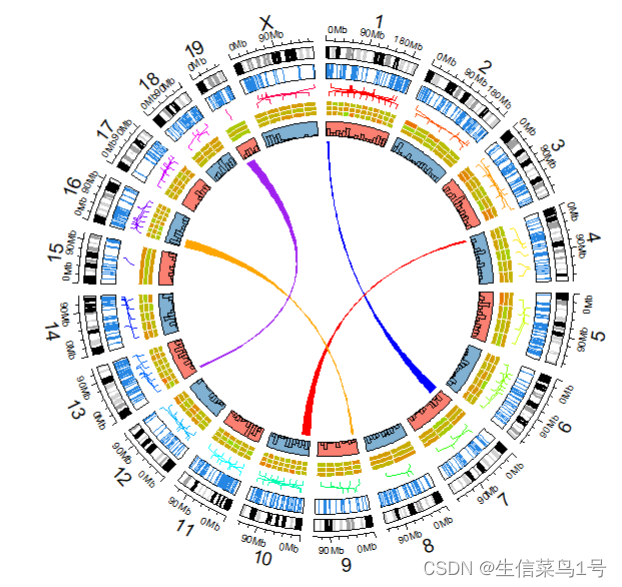
2.弦图
#直接格式 chr region chr region,h表示弧度
circos.link("chr1", c(5e6,1e7), "chr7", c(2e6,3e7), h = 0.4, col = "blue")
circos.link("chr4", c(5e6,1e7), "chr10", c(2e6,3e7), h = 0.4, col = "red")
circos.link("chr9", c(5e6,1e7), "chr16", c(2e6,3e7), h = 0.4, col = "orange")
circos.link("chr13", c(5e6,1e7), "chr19", c(2e6,3e7), h = 0.4, col = "purple")
更多绘制函数可以自行在Rstudio中了解
circos.trackxxx #大部分需要新建track,再进行绘图
circos.xxx #在circos.track()中function(x,y)类型的绘图
circos.genomic #大多为function(region,value)类型的绘图
对了,光介绍的自带数据的染色体,忘记介绍如何自行设置染色体了,等我有时间说一下,很简单。
书接上回,如果要自定义染色体的话那就不要用这个包给的物种染色体。
你需要准备一个文件,包括染色体长度名称和长度两列:
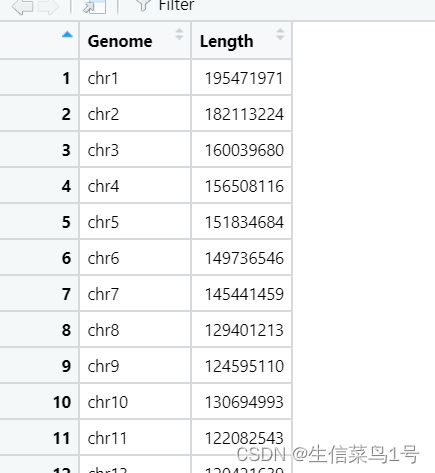
然后直接定义就好,代码如下
ref<-read.table("Genome_len.txt",header = TRUE) ## 读取染色体长度文件
circos.initialize(factors=ref$Genome, ### 定义染色体
xlim=matrix(c(rep(0,19),ref$Length),ncol=2))
这样染色体就定义好了,可以添加了track看一看
circos.track(ylim=c(0,1))
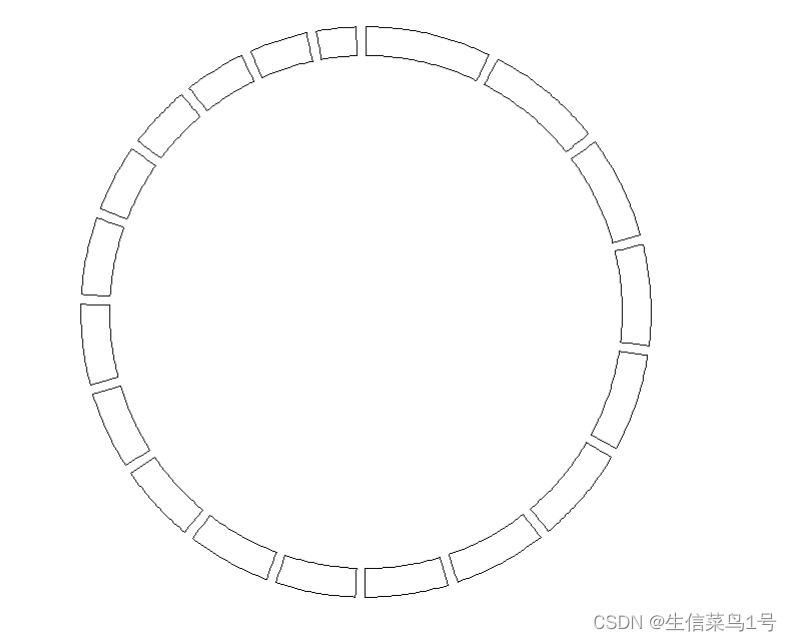
修饰一下,加上labels和染色体名称
circos.initialize(factors=ref$Genome, ### 定义染色体
xlim=matrix(c(rep(0,19),ref$Length),ncol=2))
#设置染色体框
#circos.track每条染色体按顺序轮流function,相当于一个for(chr1:chrn)循环,track.index表示绘图track的索引
circos.track(ylim=c(0,1),panel.fun=function(x,y) {
Genome=CELL_META$sector.index
xlim=CELL_META$xlim
ylim=CELL_META$ylim
#circos.rect(xlim[1], 0, xlim[2], 1, col = rand_color(19)) #跟bg.col等价
circos.text(mean(xlim),mean(ylim),Genome,cex=0.5,col=col_text,font = 2,
facing = "inside", niceFacing = TRUE)
},bg.col= rand_color(20),bg.border=F,track.height=0.06)
#定义好染色体后,在track中用CELL_META$xlim可以调用每条染色体的长度范围
#CELL_META$sector.index 染色体index
#CELL_META$ylim y的范围
brk <- seq(0,200,40)*10^6
#设置x轴axis
#track.index = get.current.track.index() 可以让轴外放
circos.track(track.index = get.current.track.index(), panel.fun = function(x, y) {
circos.axis(h="top",major.at=brk,labels=round(brk/10^6,1),labels.cex=0.4,
labels.pos.adjust =0,
col='#045eaf',labels.col='#045eaf',lwd=0.7,labels.facing="clockwise")
},bg.border=F)
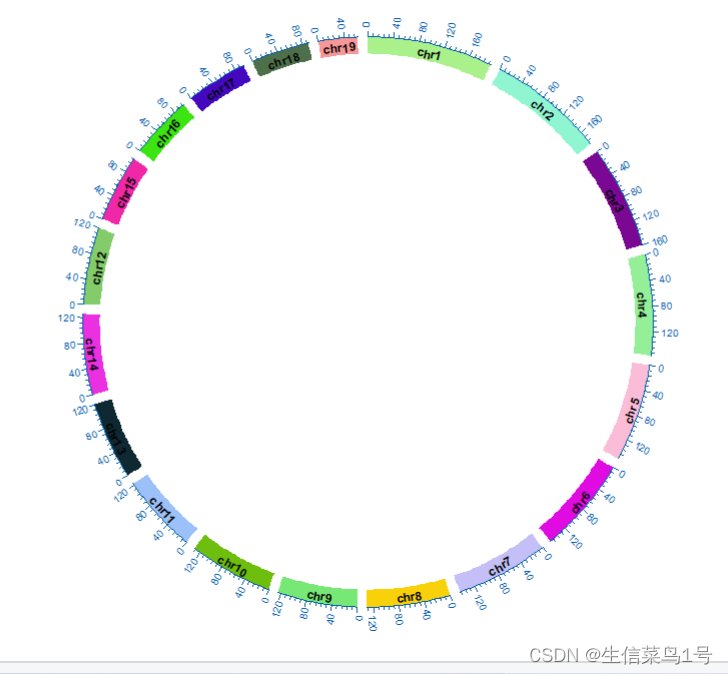
这样就可以继续进行其他图形的绘制了。

















 840
840

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









