





 本文介绍了Plink软件中BED文件的高效存储原理及其读取方法。BED文件通过位压缩技术来减少存储空间需求,每个SNP仅占用2位,极大地节省了存储空间。文中详细解释了如何使用位运算读取BED文件中的基因型数据。
本文介绍了Plink软件中BED文件的高效存储原理及其读取方法。BED文件通过位压缩技术来减少存储空间需求,每个SNP仅占用2位,极大地节省了存储空间。文中详细解释了如何使用位运算读取BED文件中的基因型数据。
Plink软件已经成为大家处理SNP的重要软件,并且大多数下游分析软件都选择将Plink格式作为输入格式。细心的朋友可能发现.bed文件相对于*.ped文件,占用存储上会小很多,bed文件是利用了什么存储原理呢?这次为大家解析一下。
正式开始之前,首先大体介绍一下计算机的存储机制,这个内容在任何C/C++书籍中应该都会提到。计算机中的存储是采用0-1形式,最小的存储单位是位(bit),相当于二进制中的一个位,而每8个位组成一个字节(Byte)。例如数字5,它在计算机中的存储形式是00000101,即1*2^2 + 0*2^1 + 1*2^0 = 5。其他存储单位的换算标准为,1KB=1024B,1MB=1024KB,1GB=1024MB,1TB=1024GB……
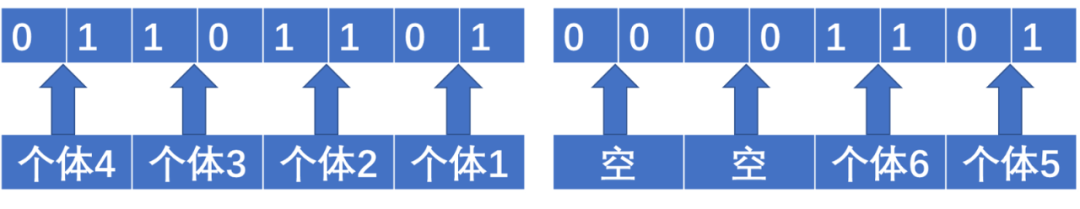
而一个个体的一个SNP位点,在存储时只占用两个bit。bed文件前三个字节存储了特定内容,从第四个字节开始存储SNP。假设我们有6个体,第四个字节是01101101。最低的两个位01表示的是第一个个体的基因型,倒数第二低的两个位11是第二个个体的基因型,再往前两个位10表示第三个个体的基因型,最高两个位01表示的是第四个个体的基因型。第五个字节的最低4位存储剩余两个个体的基因型,其余位空出来,不存储任何基因型。第二个位点从第六个字节开始存储。
两个位的0-1编码是这样表示SNP的三种基因型的:
00 Homozygous for first allele in .bim file
01 Missing genotype
10 Heterozygous
11 Homozygous for second allele in .bim file
可以看出,如果我们将基因型AA存储成C语言字符形式,需要占用2个字节,而plink存储这个基因型只需要2个位,占用空间是存储成字符型的2/(2*8)=1/8。
存储成这样的形式,我们怎么读取呢?不得不提一下位运算。这里我们用到了&(按位与)和>>(右移运算)。对于按位与运算,只有参与&运算的两个位都为1 时,结果才为 1,否则为 0。例如,10011101 & 11100001 = 10000001。对于右移运算,所有位右移若干位,高位用0补齐,例如,10011101>>2 = 00100111。
我们采用如下方式将SNP读取出来:
(1)读取一个存储SNP的字节;
(2)将这个字节与00000011(即数字3)进行&运算,最低两位为00,结果为0(纯合子);01->1(缺失);10->2(杂合子);11->3第二个纯合子;
(3)读取的字节右移两位,即将编码第二个SNP的两个位移到最低两位,进行第二步的运算;
(4)第一字节SNP提取完毕后,继续下一字节读取。
这样将bed文件提取为0、1、2、3的形式。
道理比较简单,但是操作起来可能就比较复杂了。方便读取plink bed文件,本人写了一个小程序封装到GLTA里面。使用前建议安装Anaconda(Python 3.7 version),然后利用pip install glta命令在线安装。如果以前安装过GLTA,则需要先将老版本卸载掉,命令是pip uninstall glta。
from glta.process_plink import Bed # 加载
bed_file = 'plink.qc' # plink文件
test = Bed(bed_file) # 读取文件
snp_mat = test.read()
test有四个属性:test.num_id(个体数)、test.num_snp(SNP数)、test.id_info(数据框,存储个体信息)、test.snp_info(数据框,存储SNP信息);此外还有一个方法test.read(),是转化为0、1、2、nan(缺失)后的numpy数组。
这部分程序底层是用C写的,找时间收集一下材料,做个python和C/C++混合编写的专题。欢迎大家提出建议,共同丰富GLTA软件。

















 411
411

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









