







简 介
具有复杂设计和转录分辨率分析的 RNA 测序研究涉及每个基因的多个假设;然而,传统的方法无法在基因水平上控制错误发现率 (FDR) 。提出 stageR 一种两阶段测试范式,利用聚合基因水平测试的增强能力,并允许对重要基因进行事后评估。这种方法提供了基因水平的 FDR 控制,并提高了测试相互作用效应的能力。在转录水平分析中,提供了一个框架,执行强大的基因水平测试,同时维持转录水平分辨率的生物解释。该程序适用于任何可以汇总单个假设的情况,为复杂的高通量实验提供统一的框架。

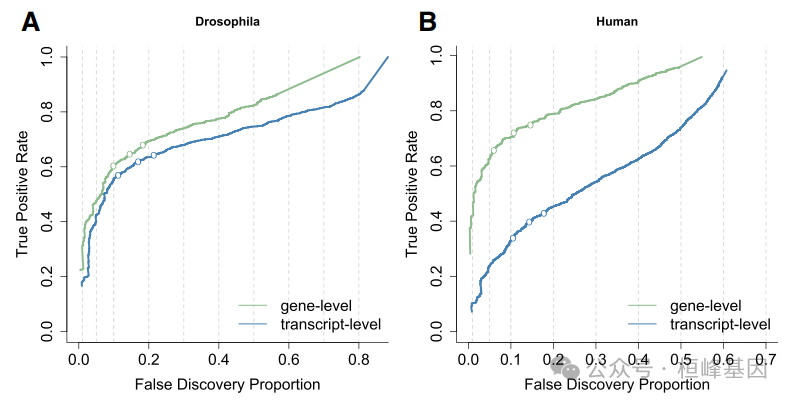
基于两次模拟数据研究的 DTU 性能曲线分析。
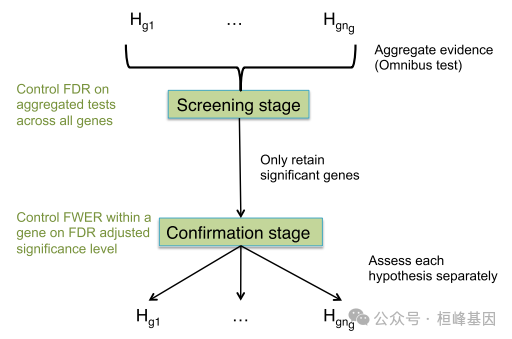
分阶段测试范式
软件包安装
软件包安装非常简单,但是需要注意以下Rtools,R,BiocManager版本号,这种问题经常出现,更新匹配一直就没问题了。
if(!requireNamespace("BiocManager", quietly = TRUE)) {
install.packages("BiocManager")
}
BiocManager::install("stageR")
#或者
library(devtools)
install_github("statOmics/stageR")加载所需要的软件包:
library(stageR)数据读取
1. 转录本差异显著的 p-values 来自 satuRn
sumExp结果来之已经准备好的结果,可以参考 RNA 44. 基于单细胞/bulk转录组的转录本差异分析(satuRn) 获得更多的详细教程。
sumExp <- readRDS("sumExp.RDS")
pvals <- rowData(sumExp)[["fitDTUResult_Contrast1"]]$empirical_pval2. 基因水平的差异显著 p-values 来自 from DEXSeq
# compute gene level q-values
geneID <- factor(rowData(sumExp)$gene_id)
geneSplit <- split(seq(along = geneID), geneID)
pGene <- sapply(geneSplit, function(i) min(pvals[i]))
pGene[is.na(pGene)] <- 1
theta <- unique(sort(pGene))
head(theta)
## [1] 4.078899e-06 1.301268e-04 1.562475e-04 2.094543e-04 2.709219e-04
## [6] 4.325295e-04
# gene-level significance testing
q <- DEXSeq:::perGeneQValueExact(pGene, theta, geneSplit)
qScreen <- rep(NA_real_, length(pGene))
qScreen <- q[match(pGene, theta)]
qScreen <- pmin(1, qScreen)
names(qScreen) <- names(geneSplit)
head(qScreen)
## ENSMUSG00000000078 ENSMUSG00000000127 ENSMUSG00000000131 ENSMUSG00000000184
## 1 1 1 1
## ENSMUSG00000000276 ENSMUSG00000000296
## 1 13. 基因与转录本ID对应列表
txInfo <- readRDS("../satuRn/txInfo.RDS")
head(txInfo)
## isoform_id gene_id
## ENSMUST00000037739 ENSMUST00000037739 ENSMUSG00000042354
## ENSMUST00000228774 ENSMUST00000228774 ENSMUSG00000042354
## ENSMUST00000025204 ENSMUST00000025204 ENSMUSG00000024346
## ENSMUST00000237499 ENSMUST00000237499 ENSMUSG00000024346
## ENSMUST00000042857 ENSMUST00000042857 ENSMUSG00000038331
## ENSMUST00000114415 ENSMUST00000114415 ENSMUSG00000038331
dim(txInfo)
## [1] 9151 24. 准备 stageR 的输入文件
tx2gene <- as.data.frame(rowData(sumExp)[c("isoform_id", "gene_id")])
colnames(tx2gene) <- c("transcript", "gene")
head(tx2gene)
## transcript gene
## ENSMUST00000037739 ENSMUST00000037739 ENSMUSG00000042354
## ENSMUST00000228774 ENSMUST00000228774 ENSMUSG00000042354
## ENSMUST00000025204 ENSMUST00000025204 ENSMUSG00000024346
## ENSMUST00000237499 ENSMUST00000237499 ENSMUSG00000024346
## ENSMUST00000042857 ENSMUST00000042857 ENSMUSG00000038331
## ENSMUST00000114415 ENSMUST00000114415 ENSMUSG00000038331
pConfirmation <- matrix(matrix(pvals), ncol = 1, dimnames = list(rownames(tx2gene),
"transcript"))
head(pConfirmation)
## transcript
## ENSMUST00000037739 0.3952020
## ENSMUST00000228774 0.4148973
## ENSMUST00000025204 0.4727593
## ENSMUST00000237499 0.4515026
## ENSMUST00000042857 0.1579658
## ENSMUST00000114415 0.1685028实例操作
1. 构建对象
准备好三个文件就可以构建stageR的对象:
# create a stageRTx object
stageRObj <- stageR::stageRTx(pScreen = qScreen, pConfirmation = pConfirmation, pScreenAdjusted = TRUE,
tx2gene = tx2gene)2. 分阶段训练
# perform the two-stage testing procedure
stageRObj <- stageR::stageWiseAdjustment(object = stageRObj, method = "dtu", alpha = 0.1,
allowNA = TRUE)3. 从stageRTx对象中检索调整后的p值
# retrieves the adjusted p-values from the stageRTx object
padj <- stageR::getAdjustedPValues(stageRObj, order = TRUE, onlySignificantGenes = FALSE)结果解读
可以看到 padj 最后基因水平校正值和转录本水平的校正值,通过 getSignificantGenes() 和 getSignificantTx() 获得显著的差异基因和差异转录本。
head(padj)
## geneID txID gene transcript
## 1 ENSMUSG00000058013 ENSMUST00000201421 0.03732585 0.05697406
## 2 ENSMUSG00000058013 ENSMUST00000201700 0.03732585 1.00000000
## 3 ENSMUSG00000058013 ENSMUST00000074733 0.03732585 1.00000000
## 4 ENSMUSG00000058013 ENSMUST00000202217 0.03732585 1.00000000
## 5 ENSMUSG00000058013 ENSMUST00000202196 0.03732585 1.00000000
## 6 ENSMUSG00000058013 ENSMUST00000202308 0.03732585 1.00000000
head(getSignificantGenes(stageRObj))
## FDR adjusted p-value
## ENSMUSG00000058013 0.03732585
head(getSignificantTx(stageRObj))
## stage-wise adjusted p-value
## ENSMUST00000201421 0.05697406Reference
Van den Berge, K., Soneson, C., Robinson, M. et al. stageR: a general stage-wise method for controlling the gene-level false discovery rate in differential expression and differential transcript usage. Genome Biol 18, 151 (2017). https://doi.org/10.1186/s13059-017-1277-0
桓峰基因,铸造成功的您!
未来桓峰基因公众号将不间断的推出单细胞系列生信分析教程,
敬请期待!!
桓峰基因官网正式上线,请大家多多关注,还有很多不足之处,大家多多指正!http://www.kyohogene.com/
桓峰基因和投必得合作,文章润色优惠85折,需要文章润色的老师可以直接到网站输入领取桓峰基因专属优惠券码:KYOHOGENE,然后上传,付款时选择桓峰基因优惠券即可享受85折优惠哦!https://www.topeditsci.com/















 2221
2221

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









