





AlphaPeptDeep: a modular deep learning framework to predict peptide properties for proteomics
期刊:Nature Communications
中科院分区:1区
影响因子:17.694
出版时间:2022 年 11 月 24 日
doi: https://doi.org/10.1038/s41467-022-34904-3
代码链接:GitHub - MannLabs/alphapeptdeep: Deep learning framework for proteomics
摘要
机器学习,尤其是深度学习 (DL) 在基于质谱 (MS) 的蛋白质组学中越来越重要。AlphaPeptDeep,这是一个构建在PyTorch DL库基础上的模块化Python框架,它可以学习和预测多肽的属性。
AlphaPeptDeep以通用的方式表示翻译后修饰,即使只有化学组成是已知的。用于预测保留时间(RT)、碰撞截面(CSS)和碎片强度的AlphaPeptDeep模型至少与现有工具相当。
其他基于序列的特性也可以通过AlphaPeptDeep进行预测,正如通过改进与数据无关的获取的HLA多肽识别模型所展示的那样
AlphaPeptDeep框架
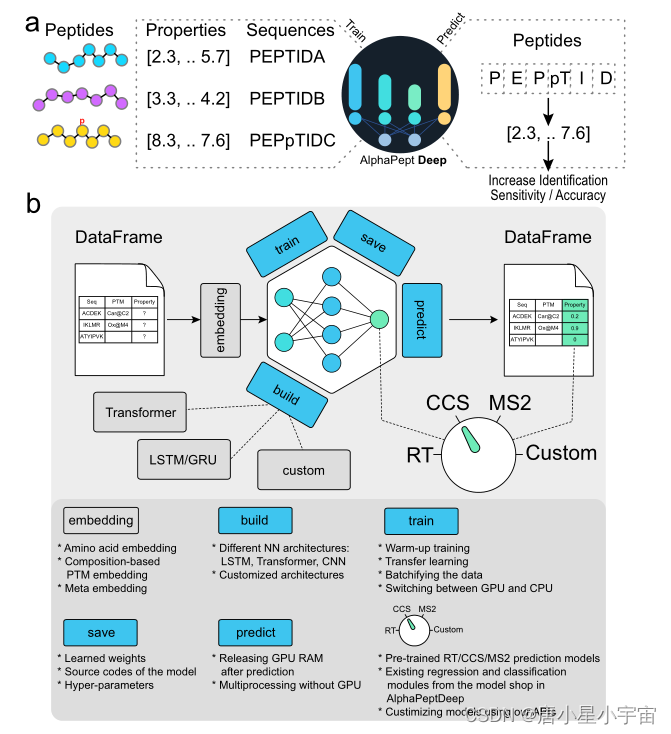
Methods
为了开发 AlphaPeptDeep,我们构建了一个名为 AlphaBase 的基础设施包,其中包含蛋白质、肽、PTM 和光谱库的许多必要功能。
在 AlphaBase 中,我们使用 pandas DataFrame 作为基础数据结构,它允许以表格格式进行数据处理。AlphaPeptDeep 使用 AlphaBase DataFrames 作为输入来构建模型并预测肽的属性。氨基酸和 PTM 嵌入直接从肽 DataFrame 中的“sequence”(氨基酸序列)、“mods”(修饰名称)和“mod_sites”(修饰位点)列执行。
Amino acid embedding
一个序列的每个氨基酸都被转换成一个唯一的整数,N 和 C 端以及其他“填充”位置使用零作为填充值。
“one-hot encoder”用于将每个整数映射到一个包含 0 和 1 的 27 维向量。使用线性层将这些向量映射到 N 维嵌入向量
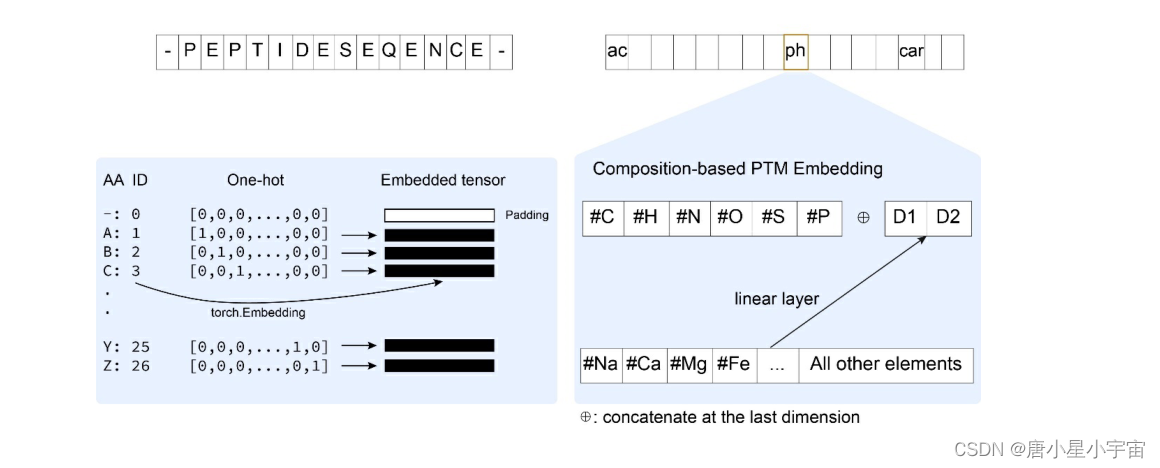
PTM embedding
对于 PTM,我们使用 6-D 嵌入向量来表示 C、H、N、O、S 和 P 原子。 PTM 的所有其他原子都嵌入到具有完全连接 (FC) 层的二维向量中。 6-D 和 2-D 向量连接成一个 8-D 向量来表示 PTM
MS2 model 、RT model、CCS model
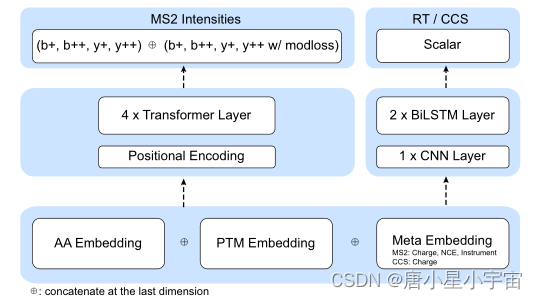
MS2 model:MS2 模型由一个嵌入层、位置编码器层和四个变换器层以及两个 FC 层组成。
嵌入层: 嵌入氨基酸序列和修饰,还嵌入元数据(如有必要),包括电荷状态、归一化碰撞能量和仪器类型。所有这些嵌入的张量都连接到下一层。
添加了一个额外的transformer层来预测“modloss”,它指的是 PTM 的中性损耗强度
RT model:RT 模型由一个用于序列和修改的嵌入层、一个 CNN 层和后面两个隐藏层大小为 128 的 Bi-LSTM 层组成。最后一个 LSTM 层的输出在肽长度维度上求和并由两个 FC 层处理输出大小为 64 和 1。
CCS model:CCS 模型包含一个用于序列、修改和电荷状态的嵌入层,以及一个 CNN 层和两个 LSTM 层,隐藏层大小为 128。最后一个 LSTM 层的输出在肽长度维度上求和并由两个 FC 层,输出大小为 64 和 1。模型参数总数为 713452。
HLA prediction model
HLA 预测模型由一个序列嵌入层、一个 CNN 层和两个隐藏层大小为 256 的 LSTM 层组成。最后一个 LSTM 层的输出在序列长度维度上求和,并由两个线性层处理,输出大小为 64 和 1。将 sigmoid 激活函数应用于最后一个线性层以获得概率。
对于训练和迁移学习,序列长度为 8 到 14 的已识别 HLA 肽被视为阳性样本。阴性样本是从与 HLA 肽具有相同长度分布的人类蛋白质序列中随机挑选的。
Results
AlphaPeptDeep 概述和模型训练
AlphaPeptDeep 框架的目标:能够轻松构建和训练深度学习 (DL) 模型,在提供足够训练数据的情况下实现高性能。
我们设计了一个通用的培训界面,允许用户只使用一行Python代码来培训模型--Model.Train()。在我们的训练界面中,我们还提供了一种“热身”训练策略(warmup),为不同的训练时期(方法)安排学习速度。事实证明,这在不同的任务中非常有用,以减少早期训练阶段的偏差。
为了构建新模型,AlphaPeptDeep 提供模块化应用程序编程接口 (API) 以使用不同的神经网络架构。(LSTM、CNN、Transformers),我们的框架还可以轻松地为不同的预测任务组合不同的神经网络架构。
AlphaPeptDeep 框架最重要的功能是预测给定目标肽的特性。仅使用 CPU 时,可以选择多处理(使用多个 CPU 内核进行预测),使得预测速度在普通个人计算机 和笔记本电脑上可以接受(整个审查的人类蛋白质组将近 2 小时)。在我们的数据集和硬件上,GPU 上的预测速度快了一个数量级。
AlphaPeptDeep 在“model shop”模块中提供了几个基于 transformers 和 LSTM 架构的模型模板来开发新的 DL 模型,并且还允许从头开始选择超参数以使用很少的代码进行分类或回归。
AlphaPeptDeep 模型对 MS2 (fragmenta-tion spectra)谱图的预测性能
第一阶段:
首先将 MS2 模型与胰蛋白酶肽数据集进行基准测试,训练和测试数据是从各种仪器和碰撞能量中收集的。
将数据集一分为二,并在类似于 pDeep 的 LSTM 模型或新的 transformer 模型上进行训练。
训练学习参数:epoch=100,warmup epoch=20,学习率 (lr)=1e–5,dropout=0.1
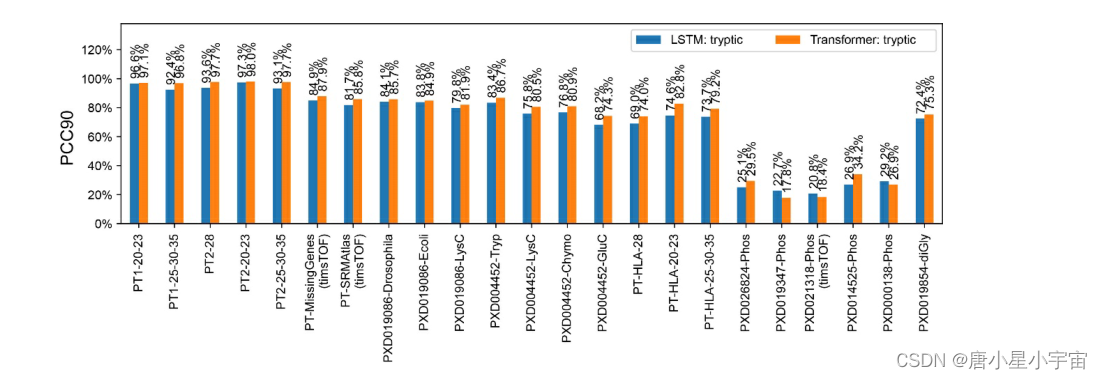
第二阶段:
将同样来自 ProteomeTools的合成 HLA 数据集附加到第 1 阶段的训练集中,并对模型进行额外 20 个epochs的训练(“微调模型”);
训练参数:epoch=20,warmup epoch=5,lr=1e–5,dropout=0.1,mini-batch size=256
第三阶段:
我们扩展了我们的模型以预测磷酸化和泛素化肽,并且只考虑局部概率 >0.75 的磷酸化位点
训练参数:epoch=20,warmup epoch=5,lr=1e–5,dropout=0.1,mini-batch size=256。
在第 3 阶段对 PTM 数据集进行训练后,性能显着提高,几乎达到胰蛋白酶肽的水平。
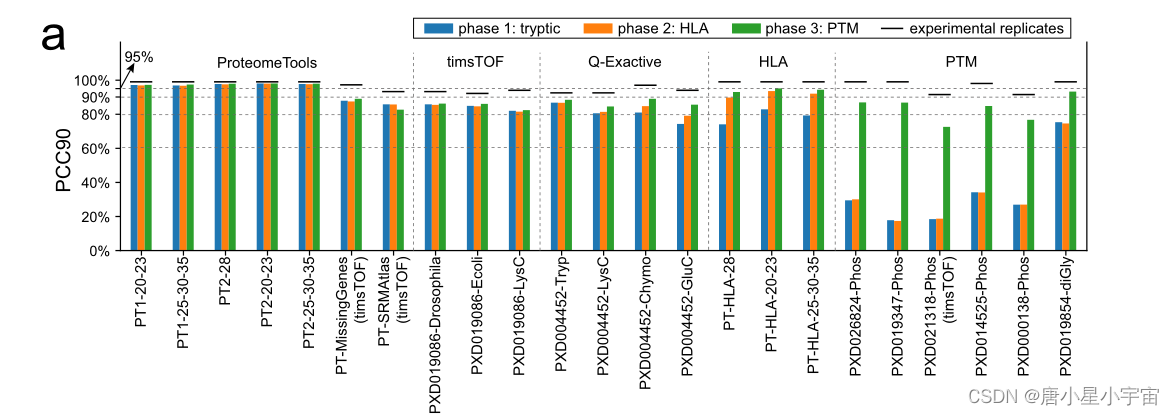
注:
- PCC90: 匹配 PSM 的预测片段强度与测量片段强度的 Pearson 相关系数 (PCC) 至少为 90%
- X轴:测试数据集的名称
- 前缀“PT”: ProteomeTools。
- PT1 和 PT2 :分别指 ProteomeTools 第 I 部分和第 II 部分。
RT (retention time)和 CCS (collision cross section)的 AlphaPeptDeep 模型的预测性能
输入:肽序列和 PTM
输出:标量值
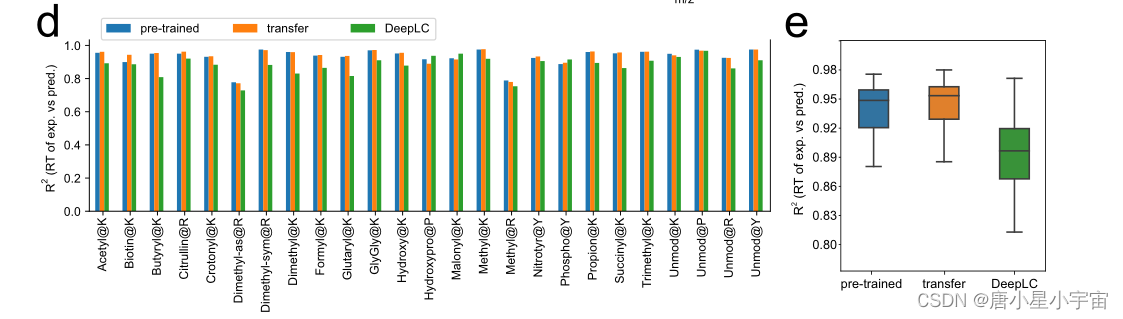
通过除以每个 LC 梯度的时间长度对训练数据集中 PSM 的所有 RT 值进行归一化,从而得到范围从 0 到 1 的归一化 RT 值。因此,预测的 RT 也被归一化。
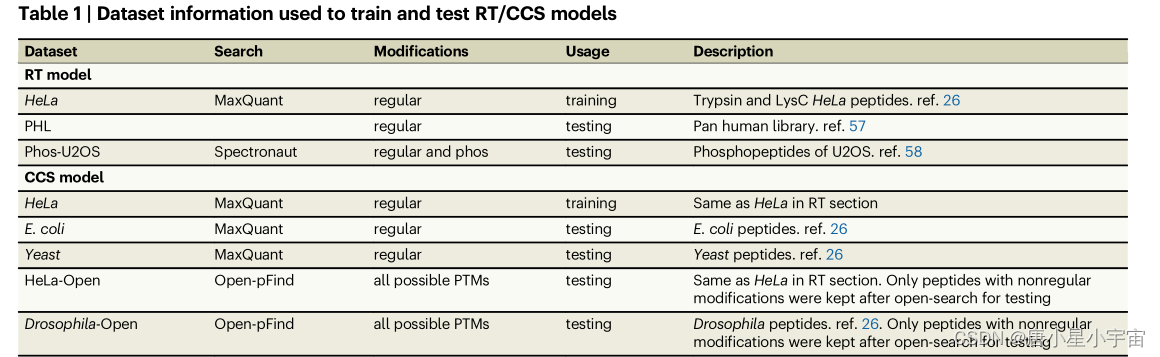
在PHL数据集中的规则肽上测试了训练好的RT模型;预先训练的模型在大部分RT范围内给出了非常好的预测,但未能准确地预测最后几分钟(对于RT预测,少样本学习可以纠正不同LC条件之间的RT偏差。)
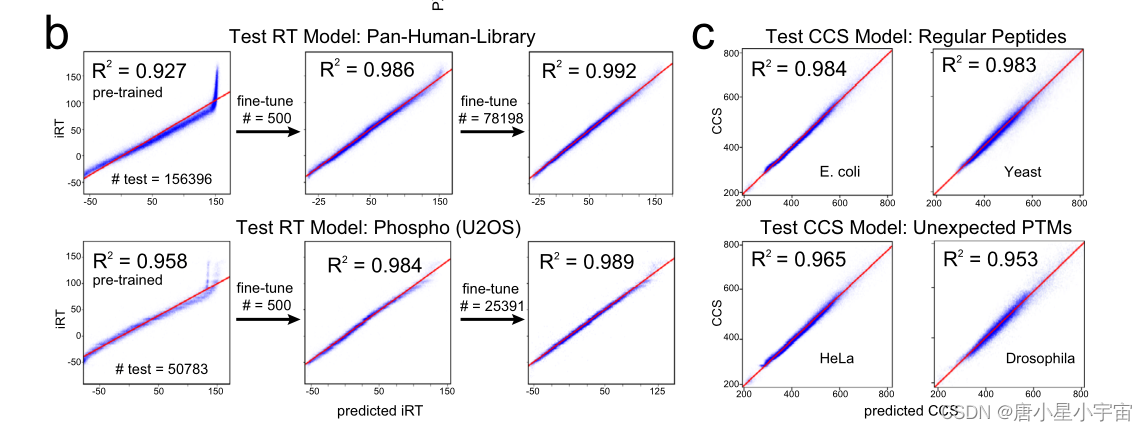
21 个带迁移学习的 PTM(翻译后修饰) 的预测性能
为了进一步证明 AlphaPeptDeep 对 PTM 的强大而灵活的支持,本文使用基于 200 个模板肽序列合成的 21 个 PTM 测试了预训练的胰蛋白酶 MS2 模型和 RT 模型;
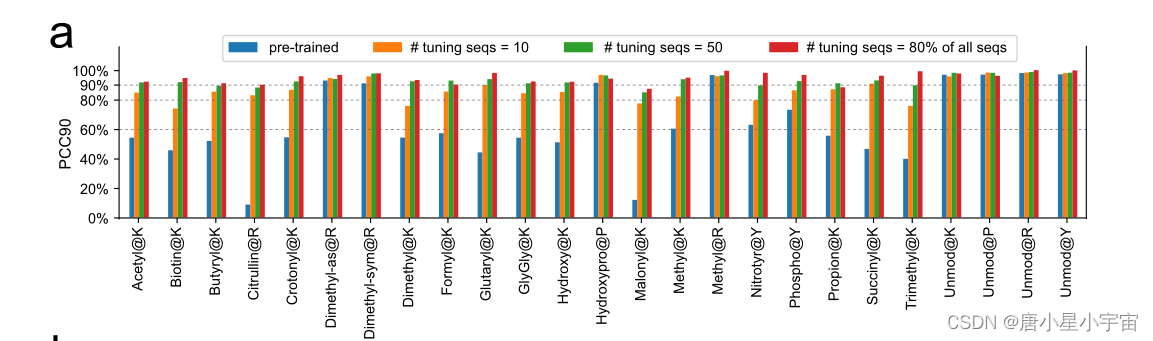
使用 10 或 50 个具有不同电荷状态和碰撞能量的训练肽对每种 PTM 类型应用迁移学习,保留具有相同 PTM 的剩余肽用于测试我们学习到的迁移;
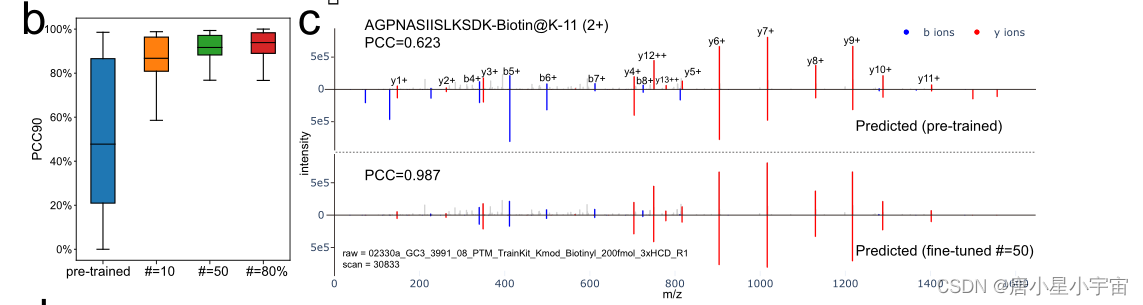
迁移学习的强大功能,我们的模型可以适应新情况,只需很少的额外数据。
使用 了21 个 PTM 的数据集测试了预训练 RT 模型的性能。尽管该模型从未在任何这些 PTM 上进行过训练,但对这些肽的 RT 预测准确性超过了 DeepLC37。
促进 HLA 肽的数据依赖采集 (DDA) 鉴定
HLA 肽是基于 MS 的蛋白质组学中最具挑战性的样品之一。鉴于 AlphaPeptDeep 中transformers出色的模型性能,假设预测 MS2 光谱可以大大提高HLA 肽的鉴定。
使用 MS2、RT 和 CCS 预测支持 HLA 肽鉴定的关键思想是,对于搜索光谱的正确肽,预测的特性应该非常接近检测到的特性,而无关肽的预测特性往往是随机分布。因此,预测和检测属性之间的相似或不同可以用作机器学习特征,以使用半监督学习来区分正确和错误的识别。AlphaPeptDeep 现在能够预测任意修饰肽的特性,甚至是具有意外 PTM 的 HLA 肽。
将 AlphaPeptDeep 结果与 MaxQuant 以及 Prosit 进行比较
在单等位基因和肿瘤数据集上,AlphaPeptDeep 覆盖了 93% 和 96% 的 MaxQuant 结果,同时在 1% 的相同 FDR 下将总数增加了一倍以上;与 Prosit 相比,AlphaPeptDeep 捕获了他们 91% 的肽,并且仍然将单等位基因数据集的总数提高了 7%。
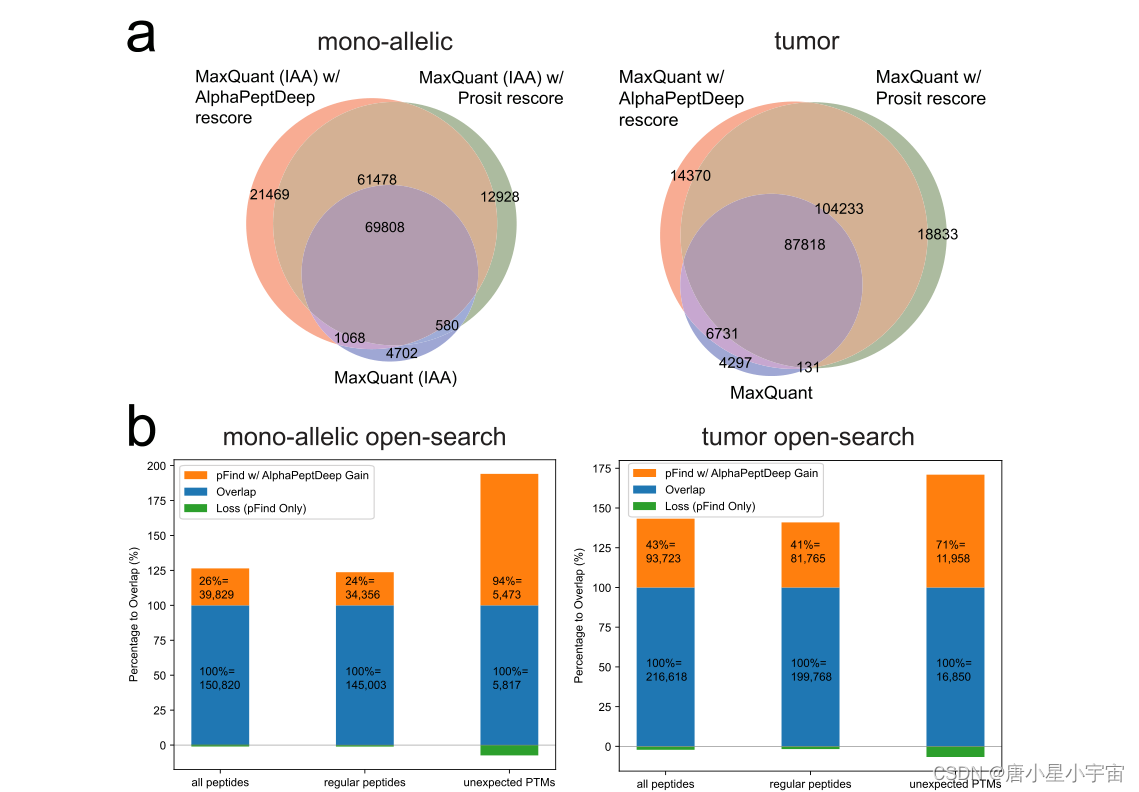
regular peptides: 指未经修饰的肽或仅经过Met-氧化和Cys-烷基化的肽
DDA: 数据依赖性采集 DIA: 数据非依赖性采集
为 HLA DIA 搜索构建 HLA 预测模型
DIA 已成为生成大规模蛋白质组数据集的首选方法。 DIA 数据分析传统上需要 DDA 实验来生成一个库,然后将数据与之匹配。
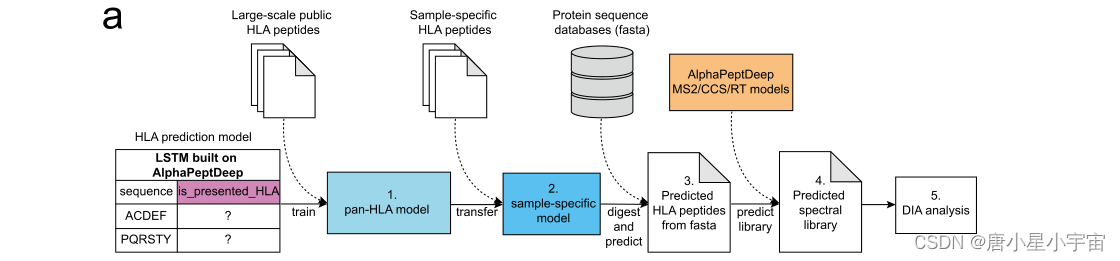
使用来自蛋白质组序列的预测 HLA library进行分析比使用实验性library进行分析更具挑战性。我们使用 AlphaPeptDeep 框架中的''model shop''功能构建了一个 HLA 预测模型。在该模型中,二元 LSTM 分类器预测给定序列是否可能是呈现给免疫系统的 HLA 肽,并从人类蛋白质组序列中提取这些肽。
该模型主要有两个目标:(1)高灵敏度 (2) 高特异性
基于这些目标,我们开发了一个模型,可以预测 DIA 数据的库搜索。分为五个步骤:
- 使用来自已知 HLA 等位基因类型的肽训练了一个泛 HLA 预测模型;(来自给定个体样本中通常存在多达 6 种不同的等位基因类型)
- 使用迁移学习来创建一个特定于人的模型,该模型具有从个体识别的特定于样本的多肽;(预测样本中是否存在潜在的HLA多肽,从而进一步减少要搜索的多肽的数量,并提高预测的准确性)
- 使用样本特定模型直接从蛋白质序列数据库中预测所有可能的个性化的HLA多肽;
- 预测的 HLA 肽用于通过使用 AlphaPeptDeep生成预测的光谱库,然后使用 DIA 搜索引擎通过 DIA 数据进行识别。
发现:在没有迁移学习的情况下直接在直接 DIA 肽上进行训练导致的肽只比迁移学习略少
推断原因:每个个体的基序不多,仅从数千个 HLA 肽中学习序列模式应该是直接的。这将有助于识别未知的样本特异性 HLA 等位基因类型,因为我们不需要任何先验知识。
但是如果我们只有少量训练肽和未知等位基因类型,迁移学习仍然是必要的。
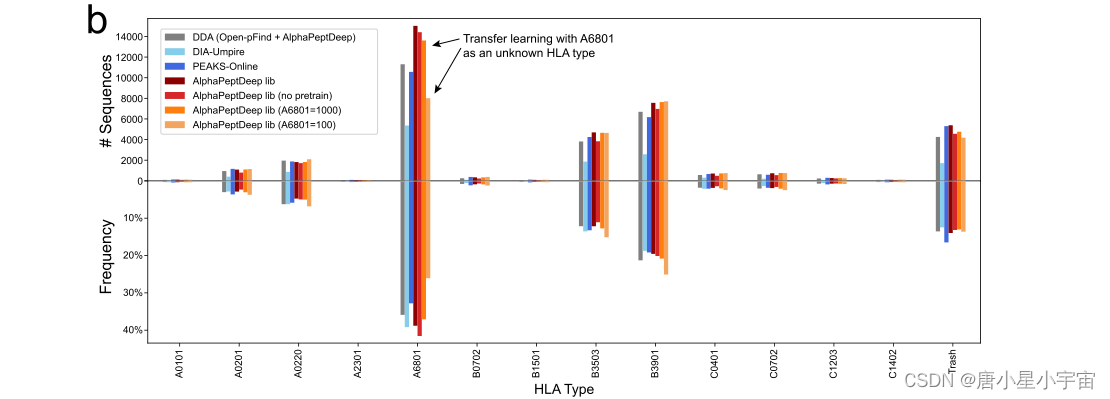
实验证明:从 94 种等位基因类型中去除了 HLA-A*68:01 的所有肽,并使用其余部分来训练新的pan- HLA 模型。这意味着样品中的所有 HLA-A*68:01 肽都不知道。然后我们仅使用 1 0 0 H L A -A * 68:01 和所有非 HLA-A * 68:01 肽,这些肽由直接 DIA 识别并由 MixMHCpred 解卷积以进行迁移学习。生成的库随后识别出 29,331 个肽段,其中 7,868 个来自 HLAA*68:01(使用 1000 个 HLA-A*68:01 肽段进行的迁移学习几乎检索到了所有肽段。
Discussion
主要工作:
- 本研究开发了一个名为 AlphaPeptDeep 的深度学习框架,它统一了高级功能来训练、迁移学习和使用模型进行肽特性预测;
- 基于这些功能,我们构建了 MS2、RT 和 CCS 模型,从而能够预测多种不同的 PTM 类型;
- 提供了一个名为“model shop”的模块,其中包含通用模型,以便用户只需几行代码即可从头开始开发新模型;
- 建立了一个 HLA 预测模型来预测一个肽序列是否是一个呈递的 HLA 肽;
亮点总结:
1.提供开源模块化、可拓展化AlphaPeptDeep框架,方便研究者直接使用。
2.通过迁移学习,可拓展AlphaPeptDeep框架多肽预测范围,满足研究者个性化需求。
目前存在的一些问题:
- 本研究直接从整个人类蛋白质组预测 HLA 谱库,并使用 HLA DIA 数据对其进行搜索。使用我们预测的库优于现有的 DDA 和 DIA 工作流程。这并不能证明 DIA 在 HLA 肽组分析中总是优于 DDA,因为 DDA 蛋白质组数据库比我们预测的 DIA 分析库大 20 倍。
- 尽管 AlphaPeptDeep 既强大又易于使用,但我们注意到传统的机器学习问题,例如框架中的过度拟合
解决方案:
- 如果未来的 DDA 搜索引擎支持预测的库搜索,它们可能能够识别更多的肽;
- 尝试不同的超参数(例如训练时期的epochs)仍然是必要的。不同的小批量大小和学习率也可能影响模型训练;
AlphaPeptDeep 能够最大限度地减少非 AI 专家的研究人员从头开始或在我们的预训练模型之上构建自己的模型的挑战。肽特性预测可以涉及几乎所有步骤,以改进计算蛋白质组学工作流程。
除了基于 MS 的蛋白质组学感兴趣的特定属性外,它原则上可用于解决肽属性是氨基酸序列函数的任何问题,正如我们通过成功预测潜在的 HLA 肽以缩小数据库搜索范围所证明的那样。因此,有了足够可靠的训练数据, AlphaPeptDeep 将可能成为蛋白质组学的宝贵深度学习资源。

















 1628
1628

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









