







今天更新TCGA数据库的利用系列第三篇文章,在对TCGA数据进行挖掘时,通常会筛选出来一些表达量显著异常的基因,作为后续研究的对象,这个筛选过程叫做差异分析;本篇文章将分为三大模块对差异分析进行介绍
关于差异分析的官方解释:
差异分析就是将一组资料的总变动量,依可能造成变动的因素分解成不同的部份,并且以假设检定的方法来判断这些因素是否确实能解释资料的变动。
我自己的一点理解:差异分析就是对总体样本数据中非正常数据的筛选。对于转录组数据进行差异分析时,limma 、edgeR、DESeq2这三种程序包都可以(limma相对较受欢迎),大部分科研性文章基本上是用其中一种方法取筛选差异表达基因,但为了使得结果更加准确,在做毕业课题时我把三种方法都做了一遍,把它们结果的交集作为筛选出来的差异表达基因。
不管用那一种方法做差异分析,基本上要做的步骤就是:一,创建表达矩阵;二、创建设计矩阵(分组矩阵);三、得到差异表达分析矩阵。
但不同包基于算法、数据模型不同,所用的函数、筛选标准也大不相同,所以用代码实现时结果有很大的差别。
无论用那种包做差异分析,在做之前必须要保证需要用到的包已经安装成功。在R语言中安装程序包的代码(其中的一种方式)如下:
source('https://bioconductor.org/biocLite.R')#加载的网址都一样
biocLite("edgeR")#把双引号的内容换成你所需要程序包的名称即可limma做差异分析
传入原始样本基因表达矩阵(表达矩阵格式如下图)
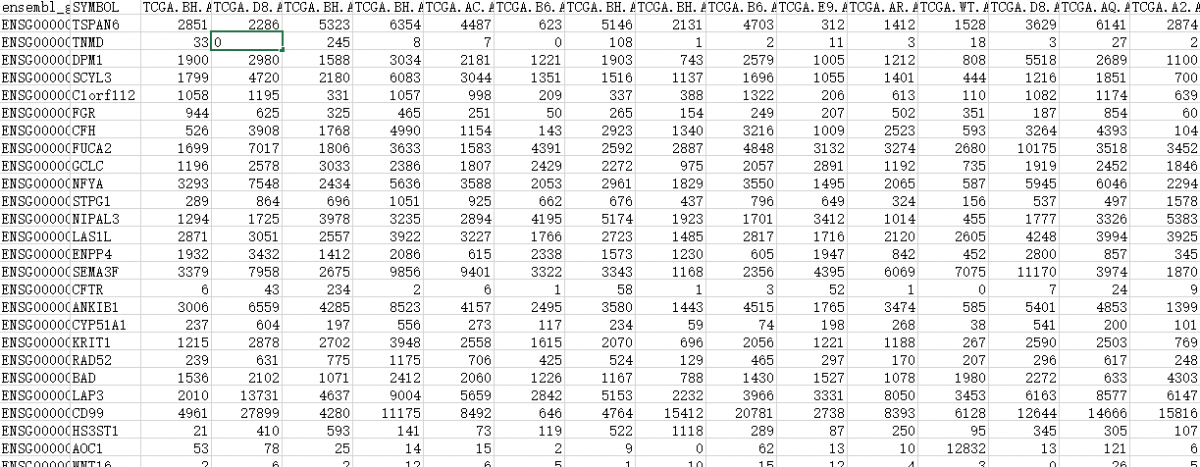
接下来就是对基因表达矩阵进行一些处理,让样本名变成数据矩阵的列名,基因名变成数据矩阵的行名,同时把ensembl_symbl那一列去掉(用express_rec <- express_rec[,(-1)]命令即可),变成如下这个格式:
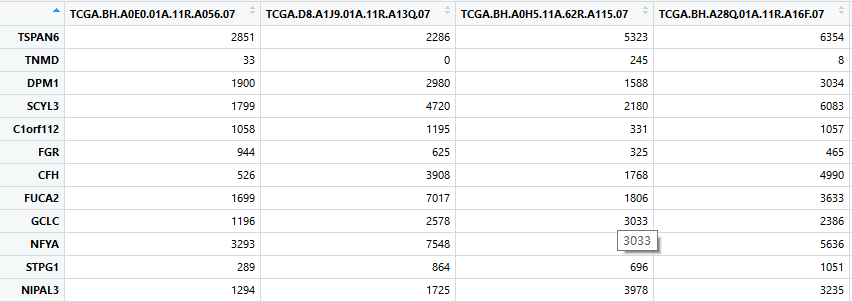
表达矩阵里面的数据太大,但为了使数据呈现正态分布,需要对数据进行标准化,这里我用的是函数log(express_rec,2),在标准化之前,需要把表达矩阵内为0的数据赋值为1,目的是为了防止取log时,数据变为负无穷大。
(express_rec[express_rec==0<-1)
下面进行分组矩阵的组建,首先提前创建好一个矩阵列表,如下,行名为样本编码,列名为样本类型,如下面这种格式:
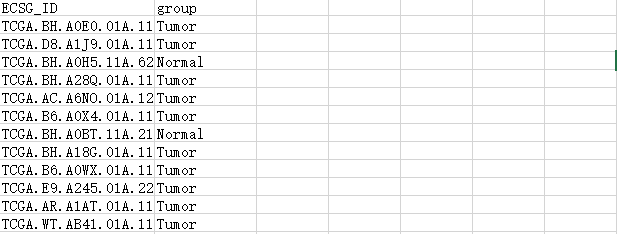
而limma包用到的设计矩阵是下面这种格式:
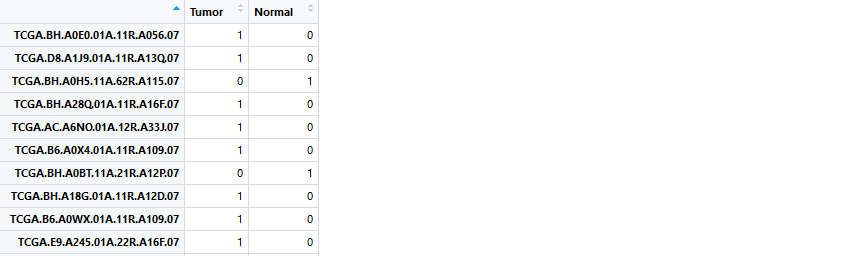
实现代码如下:
rownames(group_text) <- group_text[,1]
group_text <- group_text[c(-1)]
Group <- factor(group_text$group,levels = c('Tumor','Normal'))
design <- model.matrix(~0+Group)
colnames(design) <- c('Tumor','Normal')
rownames(design) <- rownames(group_text)
接下来的步骤依次进行数据拟合、经验贝叶斯检验、筛选差异表达基因
fit <- lmFit(express_rec,design)
#制作比对标准;
contrast.matrix <- makeContrasts(Tumor - Normal,levels=design)
fit2 <- contrasts.fit(fit,contrast.matrix)
#进行经验贝叶斯检验;
fit2 <- eBayes(fit2)
#基于logFC为标准,设置数量上限为30000,调整方法为fdr;
all_diff <- topTable(fit2, adjust.method = 'fdr',coef=1,p.value = 1,lfc <- log(1,2),number = 30000,sort.by = 'logFC')#从高到低排名;limma包的另一种方法,精确权重法(voom),然后把筛选得到的差异表达基因写入csv文件中。
#limma的另一种方法;
dge <- DGEList(counts = express_rec)
dge <- calcNormFactors(dge)#表达矩阵进行标准化;
v <- voom(dge, design,plot=TRUE)#利用limma_voom方法进行差异分析;
fit <- lmFit(v, design)#对数据进行线性拟合;
fit <- eBayes(fit)#贝叶斯算法组建
all <- topTable(fit, coef=ncol(design),n=Inf)#从高到低排名;
sig.limma <- subset(all_diff,abs(all$logFC)>1.5&all$P.Value<0.05)#进行差异基因筛选;
write.csv(sig.limma,'C:/Users/FREEDOM/Desktop/TCGA_data/limm_diff.csv')#写入csv文件中;
DESeq2做差异分析
第一步跟limma程序包一样,读入表达矩阵,对表达矩阵进行数据处理
express_rec<- read.csv('C:/Users/FREEDOM/Desktop/TCGA_data/after_note2.csv')#读取数据
group_text <- read.csv('C:/Users/FREEDOM/Desktop/TCGA_data/group_text.csv')
library('DESeq2')#加载包;
install.packages('rpart')#含有这个包可忽略,没有的时候才安装;
express_rec <- express_rec[,-1]
rownames(express_rec) <-express_rec[,1]
express_rec <- express_rec[(-1)]#表达矩阵的处理;
rownames(group_text) <- group_text[,1]
创建分组矩阵
rownames(group_text) <- group_text[,1]
group_text <- group_text[c(-1)]#分组矩阵的数据处理;
all(rownames(group_text)==colnames(express_rec))#确保表达矩阵的列名与分组矩阵行名相一致;
构建 DESeq2 所需的 DESeqDataSet 对象
dds <- DESeqDataSetFromMatrix(countData=express_rec, colData=group_text, design<- ~ group) #DESeq2的加载
head(dds)
dds <- dds[rowSums(counts(dds)) > 1, ] #过滤一些low count的数据;
使用DESeq进行差异表达分析,返回 results可用的DESeqDataSet对象
> dds <- DESeq(dds)#DESeq进行标准化;
estimating size factors
estimating dispersions
gene-wise dispersion estimates
mean-dispersion relationship
final dispersion estimates
fitting model and testing
-- replacing outliers and refitting for 2819 genes
-- DESeq argument 'minReplicatesForReplace' = 7
-- original counts are preserved in counts(dds)
estimating dispersions
fitting model and testing
可以用summary函数查看表达基因上下调分布基本情况
> summary(res)#查看经过标准化矩阵的基本情况;
out of 24823 with nonzero total read count
adjusted p-value < 0.1
LFC > 0 (up) : 7590, 31%
LFC < 0 (down) : 5120, 21%
outliers [1] : 0, 0%
low counts [2] : 0, 0%
(mean count < 0)
[1] see 'cooksCutoff' argument of ?results
[2] see 'independentFiltering' argument of ?results
最后提取差异表达基因,存入csv文件中。
edgeR进行差异分析
这个包的操作步骤与limma包类似,首先就是读入数据,创建表达、分组矩阵。
path = 'C:/Users/FREEDOM/Desktop/TCGA_data/after_note2.csv'
path1 ='C:/Users/FREEDOM/Desktop/TCGA_data/group_text.csv'
express_rec <- read.csv(path,headers <- T)#读取表达矩阵;
group_text <- read.csv(path1,headers <- T)#读取分组矩阵;
library(edgeR)#加载edgeR包
express_rec <- express_rec[,-1]
rownames(express_rec) <- express_rec[,1]
express_rec <- express_rec[(-1)]#创建表达矩阵;
rownames(group_text) <- group_text[,1]
group_text <- group_text[c(-1)]#加载分组矩阵;
group <-factor(group_text$group)
dge <- DGEList(counts = express_rec,group = group)#构建DEList对象;
y <- calcNormFactors(dge)#利用calcNormFactor函数对DEList对象进行标准化(TMM算法)
#创建设计矩阵,跟Limma包相似;
Group <- factor(group_text$group,levels = c('Tumor','Normal'))
design <- model.matrix(~0+Group)
colnames(design) <- c('Tumor','Normal')
rownames(design) <- rownames(group_text)#创建分组矩阵;
edgeR包创建的分组矩阵与limma一样,是以factor因子格式展现出来

接下来依次进行构建DGEList对象、利用TMM算法对数据进行标准化、估计离散值、数据拟合、创建对比矩阵、对数据做QL-text检验、差异表达基因写入csv文件中
dge <- DGEList(counts = express_rec,group = group)#构建DEList对象;
y <- calcNormFactors(dge)#利用calcNormFactor函数对DEList对象进行标准化(TMM算法)
y <- estimateDisp(y,design)#估计离散值(Dispersion)
fit <- glmQLFit(y, design, robust=TRUE)#进一步通过quasi-likelihood (QL)拟合NB模型
TU.vs.NO <- makeContrasts(Tumor-Normal, levels=design)#这一步主要构建比较矩阵;
res <- glmQLFTest(fit, contrast=TU.vs.NO)#用QL F-test进行检验
# ig.edger <- res$table[p.adjust(res$table$PValue, method = "BH") < 0.01, ]#利用‘BH’方法;
result_diff <- res$table#取出最终的差异基因;
write.csv(edge_diff,'C:/Users/FREEDOM/Desktop/TCGA_data/edgeR_diff2.csv')
以上就是做差异分析三种R包的使用方法,关于本篇文章涉及到的完整源码的获取方式:关注公众号:小张Python,后台回复关键词:差异分析 即可。
















 2690
2690











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











