







作者:佳名来源:简书 - R 语言文集
1. 读取并处理基因表达数据
这是我的基因表达量数据:
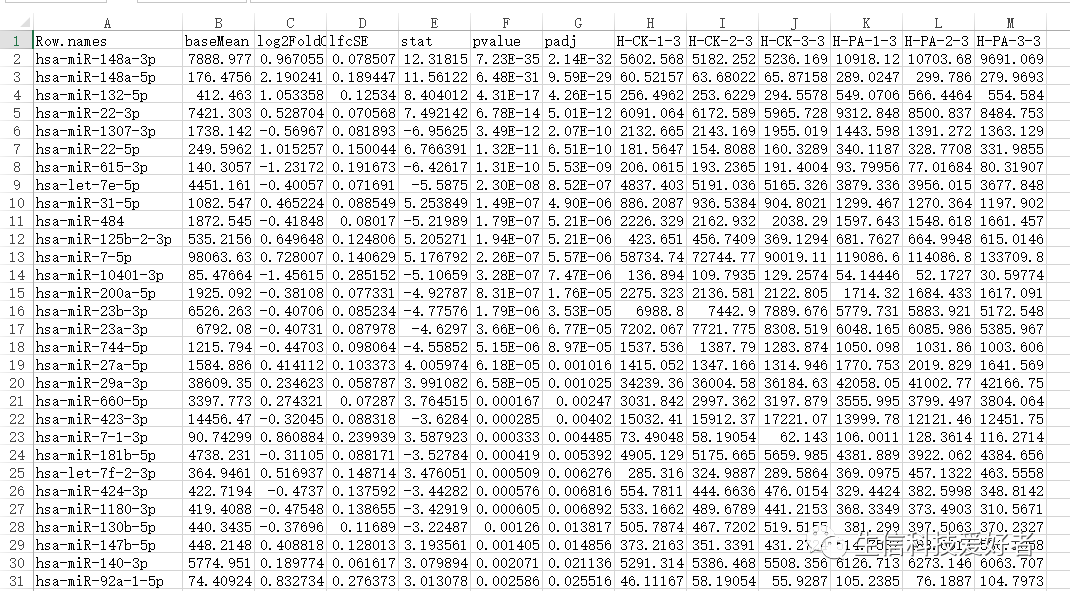
> myfiles "*.csv")
> myfiles
[1] "4_total_total_FPKM.csv"
> matrix 1], sep=',', header=T, check.names=FALSE, row.names=1)
1.1 提取部分数据集
提取 padj 的值小于 0.2 的数据:
> matrix 0.2)
1.2 提取基因表达值所在的列,组成新的矩阵,并将矩阵转置
由于 R 语言的 scale 函数是按列归一化,对于我们一般习惯基因名为行,样本名为列的数据框,就需要进行转置。
mat 7:12]) # 7-12 列为每个样本的基因表达量
1.3 基因表达归一化
mat TRUE, scale = TRUE)
View(mat)
1.4 对数据进行聚类,从而得到其 dendrogram
# dist 函数计算 microRNA 间的距离, hclust 函数用来进行层次聚类.
dend
1.5 定义进化树颜色
library(dendextend)
n 3 # n 可自定义
dend % set("branches_k_color", k = n)
1.6 可视化处理
par(mar=c(7.5,3,1,0))
plot(dend)

1.7 聚类后的矩阵
如图 Fig 3,聚类后的矩阵的列的顺序会发生变化。按此顺序,重新排列矩阵。
mat2  最低0.47元/天 解锁文章
最低0.47元/天 解锁文章
















 5574
5574











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









