






绘制基因组水平的热图是一个非常普遍的需求,比如下图(图片来源于https://doi.org/10.1101/594267)
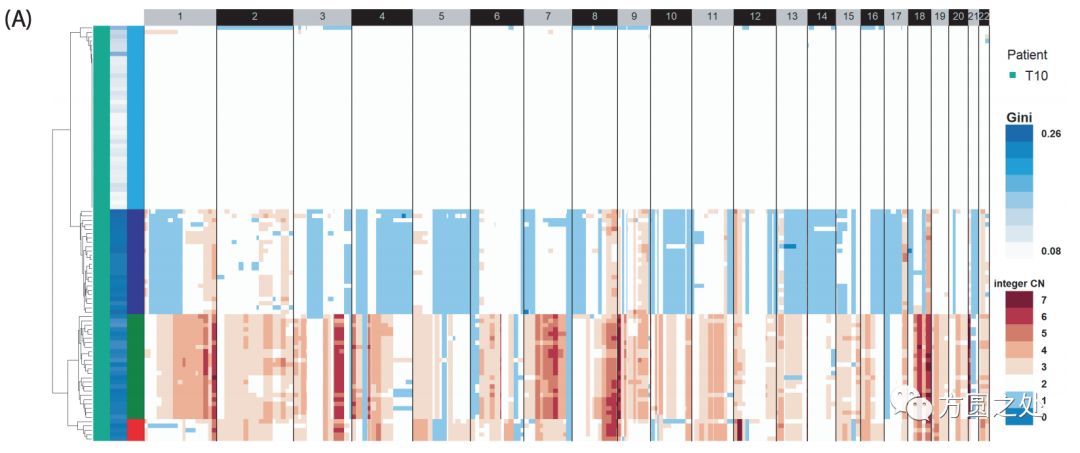
在这种热图中,行或者列的顺序对应着在基因组中的位置,并且以染色体进行分组和排序。在这篇文章中,我将介绍如何使用ComplexHeatmap包绘制基因组水平的热图,同时基于ComplexHeatmap强大的功能,也可以在热图上添加更多的元素,比如复杂的annotation track。
由于基因组中动辄上百万到上十亿的碱基,我们不可能对每个碱基进行可视化,核心的思路是将原始基因组划分为几百个或者几千个长度相同的window,然后把所有相关的基因组数据转换成对应于这些window的数值。
从ComplexHeatmap版本2.7.1.1003开始,我加入了两个函数bin_genome()和normalize_genomic_signals_to_bins()用来对基因组进行划分和对基因组水平的数据进行转换,从而生成合适的矩阵用来进行热图的可视化。在这里我简单介绍下这两个新函数的使用方法。
在下文中,我们使用术语“genomic windows”来表示基因组被切分后的长度相同的region,使用“genomic signals”来表示将要被可视化的基因组数据,例如,DNA甲基化值或者CNV突变数据。
bin_genome()的使用方法十分简单和直接。你只需要设定基因组和genomic windows的大小,你也可以设定需要使用哪些染色体。
library(ComplexHeatmap)
chr_window = bin_genome("hg19")
chr_window
## GRanges object with 1990 ranges and 0 metadata columns:
## seqnames ranges strand
##
## [1] chr1 1-1547839 *
## [2] chr1 1547840-3095678 *
## [3] chr1 3095679-4643517 *
## [4] chr1 4643518-6191356 *
## [5] chr1 6191357-7739195 *
## ... ... ... ...
## [1986] chrY 51078688-52626526 *
## [1987] chrY 52626527-54174365 *
## [1988] chrY 54174366-55722204 *
## [1989] chrY 55722205-57270043 *
## [1990] chrY 57270044-58817882 *
## -------
## seqinfo: 24 sequences from an unspecified genome; no seqlengths
返回值chr_window是一个GRanges对象,其中包含着基因组被切分成后的genomic windows的位置。这个返回值并不重要,因为这些genomic windows已经被内部保存了(将在下文中解释)。
在bin_genome()中有一些参数可能需要被注意:
species:基因组的缩写,比如"hg19"和"mm10"。bins:基因组被切分的数目。最终的genomic windows的数据大致近似于这个值。bins其实是用来计算一个合适的bin_size值。bin_size:window的大小。如果这个参数被设定,bins参数将被忽略。...:所有其他的参数都被传递到circlize::read.chromInfo()中。如果你的基因组没有合适的缩写的话,你可以构建一个数据框,其中包含着每条染色体的起始位置和终止位置,然后传递给chromInfo参数,或者你也可以通过设定chromosome.index来选择一部分染色体。
第二个函数normalize_genomic_signals_to_bins(),正如函数名所表示的,这个函数用来把基因组水平的数据(genomc signals)对应到每一个genomic window中去。在如下例子中,
x = normalize_genomic_signals_to_bins(gr,
value, value_column, method, empty_value)
gr是包含了genomic signals的在基因组中的位置。对每一个window,normalize_genomic_signals_to_bins()会计算genomic signals相交到此window的平均值。normalize_genomic_signals_to_bins()返回一个矩阵,其拥有和genomic windows相同数目的行。在多次执行normalize_genomic_signals_to_bins()返回的矩阵中,它们的行都相互对应,也就是所有矩阵中的第i行都对应着第i个genomic window,这也是为什么函数操作叫”normalize“的原因。
normalize_genomic_signals_to_bins()函数中的参数有:
gr:genomic signals在基因组中的位置,必须是一个GRanges对象。value:gr所对应的数值,可以是一个向量或者矩阵,行必须与gr相对应。value_column:如果gr对应的数值已经保存在gr的meta column中,那么可以通过设定value_column来选择指定的列。method:将genomic signals平均到genomic windows中的方法,一共有三种选择:"weighted","w0"和"absolute"。我将在下面详细介绍。empty_value:如果没有任何基因组信号与window重合,这是这些window的缺省值。
下图展示了ovelapping model,其中下端的红色线段表示某一个genomic window,黑色线段表示不同的genomic signals,粗线段表示和window重叠的部分。这五条线段都有一个值对应,我们记为,,,和。
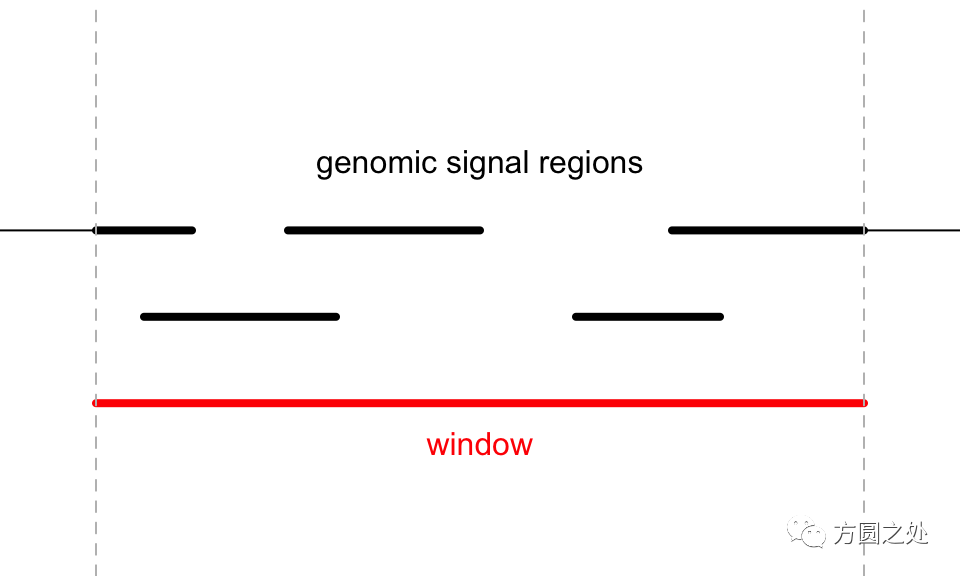
对于某一个window,记为和此window相交的genomic signals的个数(在示意图中为5),是与此window相交的genomic region的长度(也就是黑色粗线段),是这些signal region所对应的值。
如果基因组信号是数值型的,表示为一个向量或者一个矩阵,那么有三种方法:
absolute方法表示为,简单的定义为的均值。注意在此,genomic signal region的长度将被忽略:
weighted方法表示为,定义为以signal region长度作为权重的的均值:
当基因组背景值不需要考虑的情况下,可以使用"absolute"和"weighted" 方法,比如说当我们把DNA甲基化数据平均到每一个window中时,我们不需要考虑非CpG的位点,因为甲基化只和CpG相关。
w0方法考虑了背景值,它是和signal region相交部分和不相交部分的均值:
其中是相交部分的长度和,是不相交部分的长度和。
如果基因组信号是字符型向量或者离散向量,当前window所赋予的值是与此window相交的拥有最长的signal region的水平值。我们把离散向量所有的水平记为,的某一个水平记为,那么最终赋予此window的值为:
除了这些预定义的overlapping的方法以外,用户还可以自定义自己的overlap的方法。用户可以提供一个自定义函数给method参数。此函数接受三个参数 x,w和gw。此函数将被作用到每一个window中。此函数的三个参数为:
x:和当前window相交的signal region所对应的值。也就是示意图中的.w:和当前window相交的signal region的宽度。也就是示意图中粗黑色线段的长度。gw:当前window的长度。
此函数必须返回一个单独值(a scalar)。那么,"weighted"方法可以被定义为:
function(x, w, gw) sum(x*w)/sum(w)
normalize_genomic_signals_to_bins()函数允许下面格式的value:
- 当
value或者value_column都没有提供时,此函数简单的overlapgr和genomic windows,返回一个一列的logical的矩阵,其中的值表示当前的window是否和gr有overlap。 - 当基因组信号是连续数值型的,
value可以是一个向量或者一个矩阵,如果信号值已经在gr中了,value_column可以是一个单独的值或者多个列的index。此函数返回一个数值型的矩阵。 - 当基因组型号是离散型或者字符型的,
value只能是一个向量,或者value_column只能包含一个单独的值。此函数返回一个一列的字符型的矩阵。
在使用normalize_genomic_signals_to_bins()函数时,用户不需要提供genomic windows变量。当bin_genome()被执行之后,genomic windows变量会被内部保存,然后以后每一次使用normalize_genomic_signals_to_bins()都会直接使用这个内部保存的genomic windows变量。这样有一个好处,对不同的genomic signals进行多次运行normalize_genomic_signals_to_bins(),它总是能返回相同行的矩阵。这样就可以安全的对多个heatmap进行连接了。
这基本上就是全部了,normalize_genomic_signals_to_bins()返回normalize好了的矩阵,然后你可以直接在Heatmap()或者相关的热图函数中使用。
下图绘制了一个复杂的基因组水平的热图,其中我也加入了更多的annotaiton track。详细代码请见“阅读原文”。
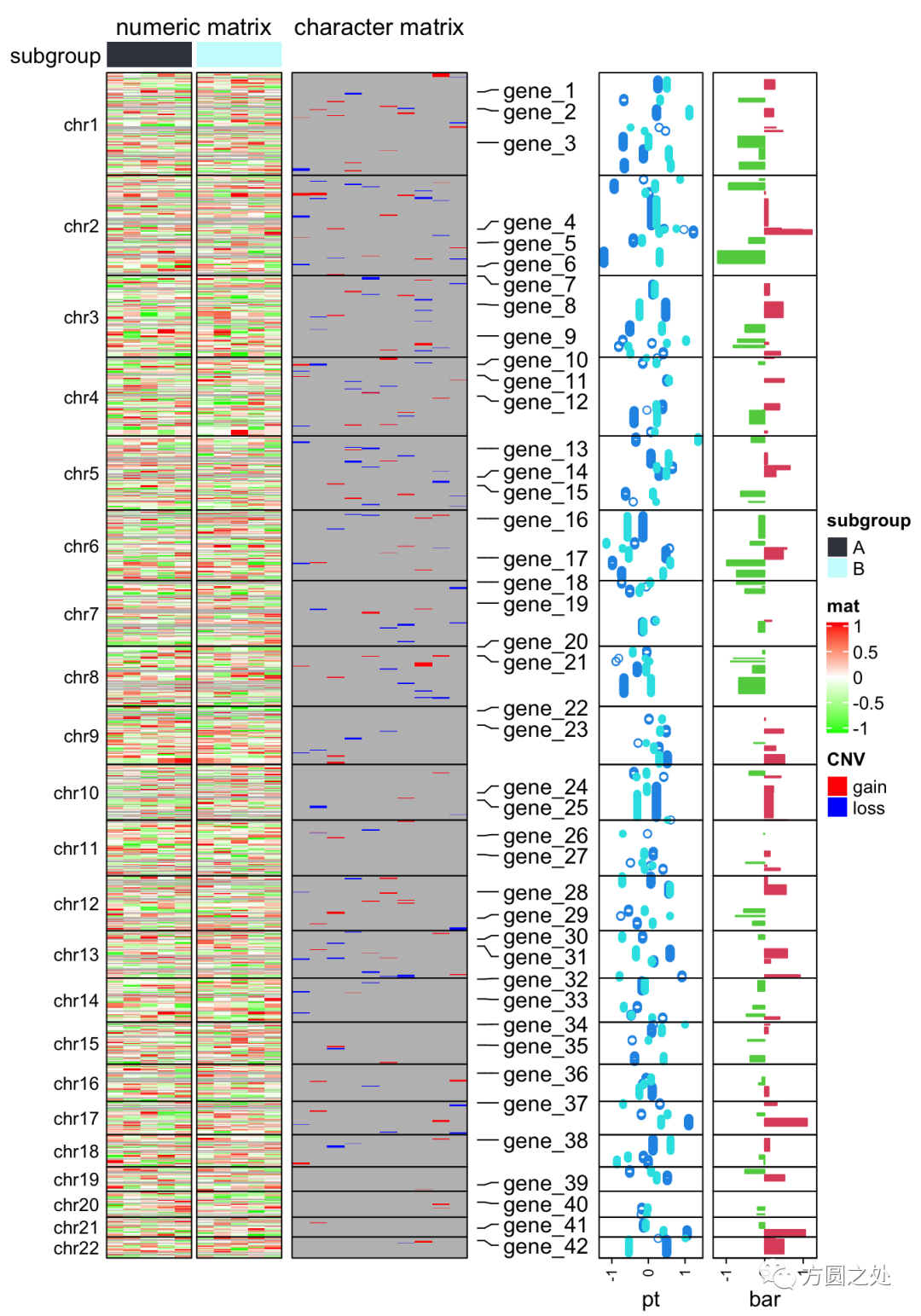
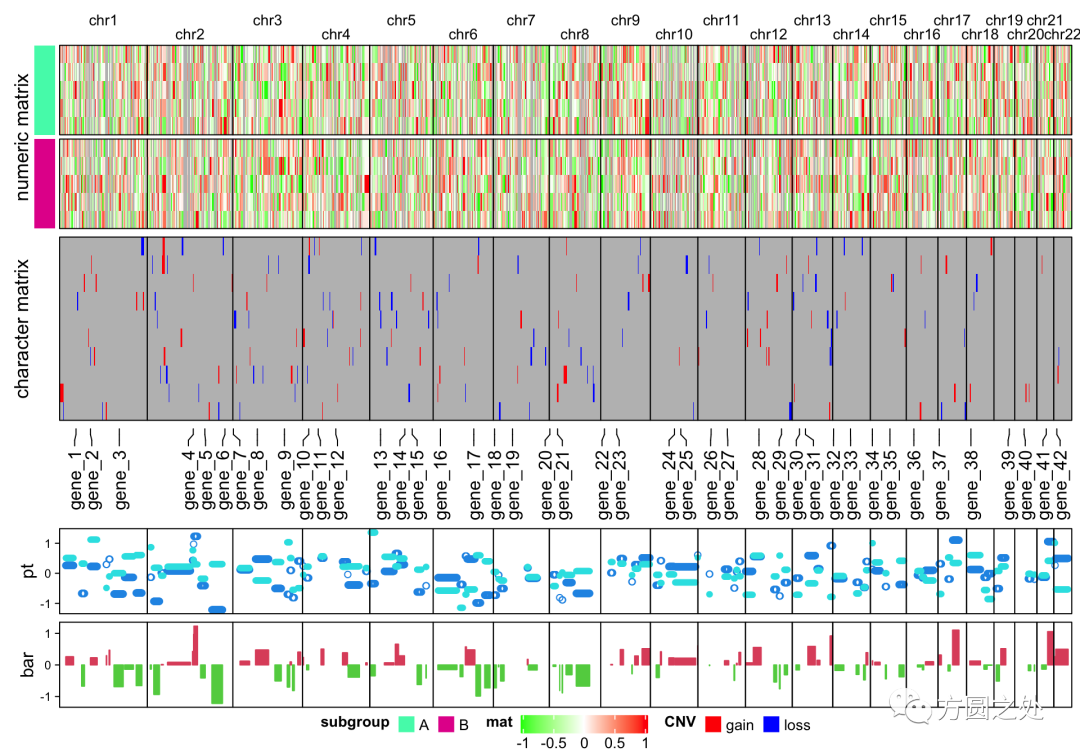
在上图中,染色体是垂直分布的。在ComplexHeatmap中,可以很简单将其翻转成水平方向(只需将行和列的设置互换,使用%v%连接符)。详细代码请见“阅读原文”。















 902
902











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









