







欢迎大家关注 微信公众号|计算生物前沿
近期,《Nature Communications》发表了一项名为NuFold的突破性研究,提出了一种端到端深度学习模型,用于高效预测RNA的三级结构。该方法通过创新的“核碱基中心表示”技术,克服了传统方法在RNA局部几何结构建模上的不足,性能显著优于基于能量的方法,并与现有深度学习方案相媲美,尤其在局部结构准确性上表现突出。

引言
研究背景与现状:RNA作为遗传信息载体和功能分子(如核酶、非编码RNA),其三维结构直接决定生物学活性,但实验解析面临严峻瓶颈——PDB数据库中仅3%为RNA结构(约6000个),且X射线、冷冻电镜等方法耗时耗力。现有计算工具亦存在局限:模板法依赖同源结构导致适用性狭窄,能量优化法精度低且计算效率不足,深度学习方法(如DeepFoldRNA)仍需后处理折叠步骤,无法直接输出原子模型。
不足与挑战:RNA结构预测的挑战源于其核糖环的灵活构象(如C3’-endo与C2’-endo)对局部几何的精准建模需求,而实验数据稀缺更制约了深度学习模型的泛化能力。尽管已有端到端模型(如RhoFold)尝试突破,但其对二级结构整合不足,且未优化核碱基旋转自由度,导致建模断层问题显著。
文章目标:提出首个端到端全原子RNA结构预测工具NuFold,直接从序列输出三维原子模型,核心创新包括:通过核糖环扭转角预测实现局部构象精准控制,整合元基因组数据增强与自蒸馏策略扩展训练集,并采用动态循环迭代优化提升长RNA建模精度,突破传统多步骤方法误差累积的瓶颈,为RNA药物研发提供全新解决方案。
NuFold方法概述
基于AlphaFold2架构改造,输入RNA序列,通过深度网络直接预测全原子坐标,结合多序列比对(MSA)与二级结构信息迭代优化。
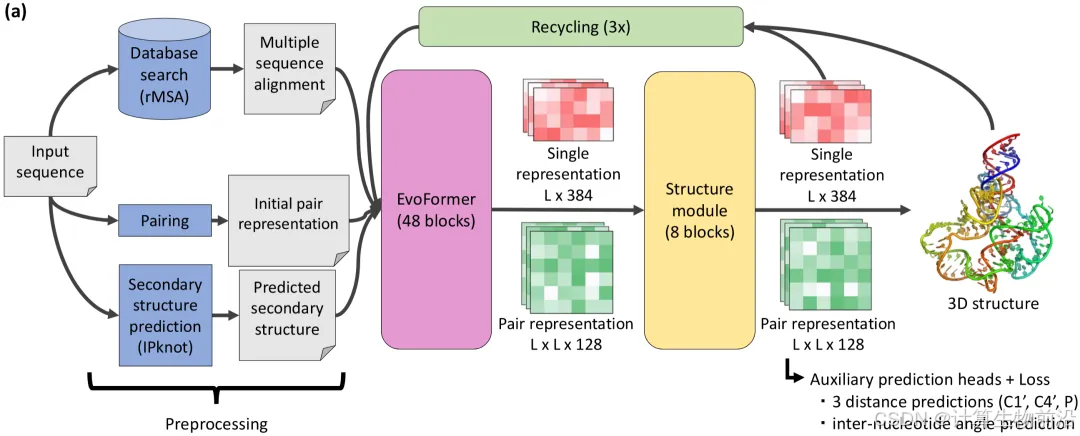
核心流程包括:
输入预处理
-
多序列比对(MSA):利用rMSA从公共数据库(RNAcentral、NCBI nt等)检索同源序列,并通过元基因组数据扩增多样性。
-
二级结构预测:IPknot预测碱基配对信息,提供局部相互作用约束。
特征提取(EvoFormer模块)
-
从MSA中提取共进化信号,构建残基对距离与角度分布。
-
二级结构信息嵌入单残基特征,强化局部几何建模。
结构模块与循环迭代
-
灵活核碱基表示:以C1'原子为基准点,预测四原子基架的位置及核糖环扭转角,逐层构建全原子模型。
-
循环优化(Recycling):输出结构反馈至输入,经3~30次迭代逐步修正。
自蒸馏数据增强
利用trRosettaRNA预测未标注序列(bpRNA-1m数据集)生成伪标签,扩展训练数据量。
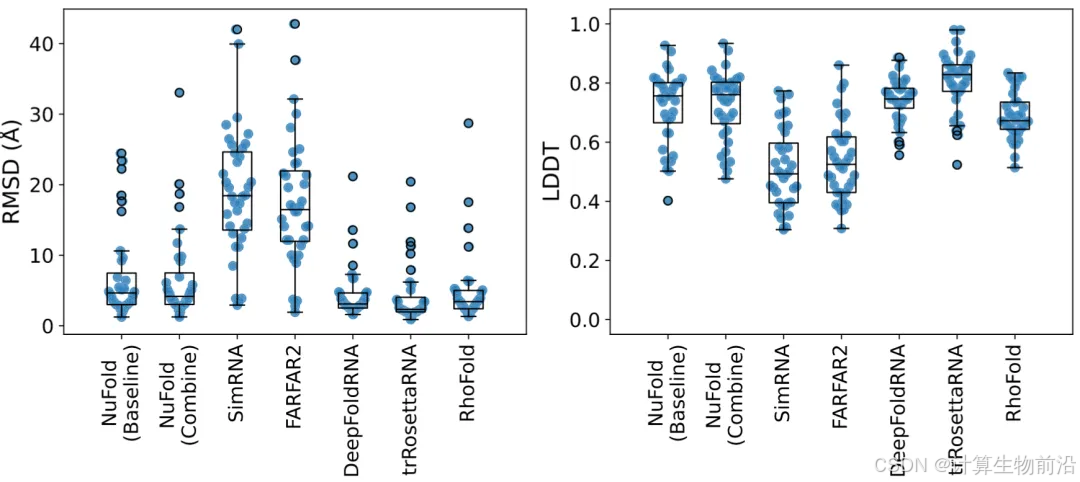
NuFold可以精准建模RNA结构
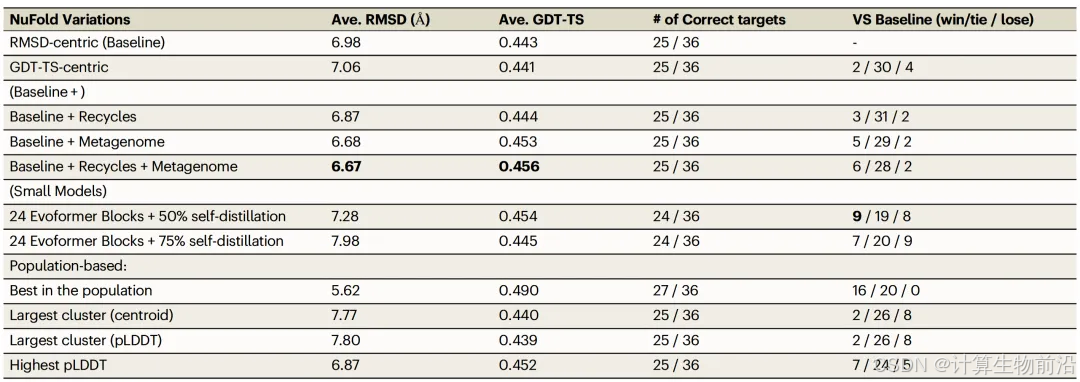
NuFold在36个RNA测试集上展现了显著优势,平均C1'原子RMSD为6.98 Å,其中25个结构(69%)预测误差低于6 Å。
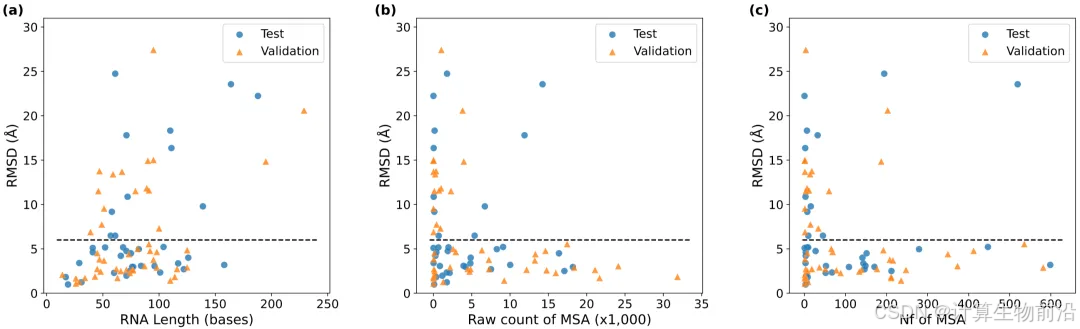
模型具有目标长度与MSA深度的依赖性
短RNA(<50 nt)预测准确率高(均低于6 Å),而长RNA(>100 nt)更依赖深度MSA。当有效序列数(Nf)低于100时,39.3%的案例误差超过6 Å。
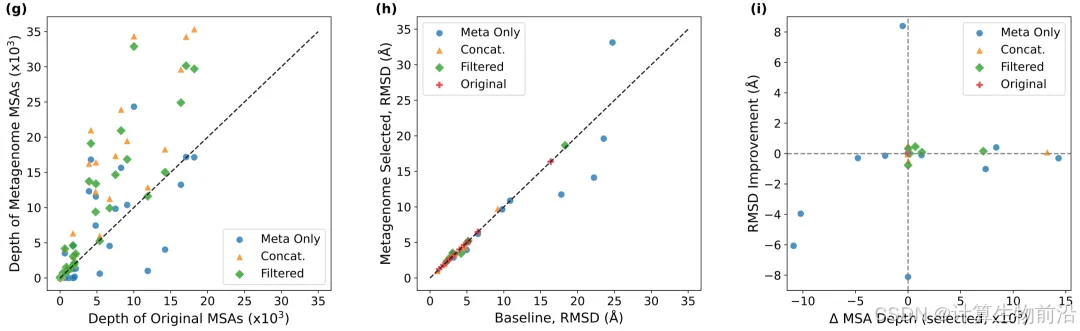
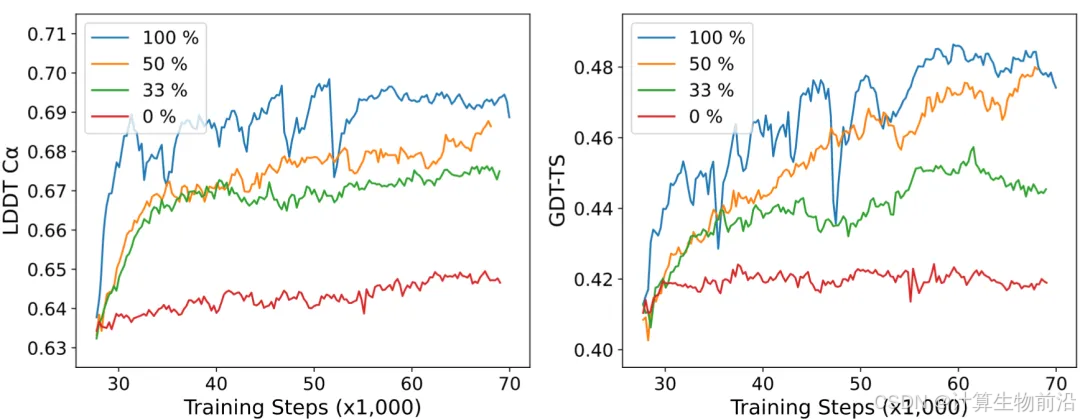
元基因组与循环迭代会促进模型性能
引入元基因组扩增MSA,使34/36案例精度提升(平均RMSD降0.3 Å);结合10次循环优化后,平均RMSD进一步降至6.67 Å。
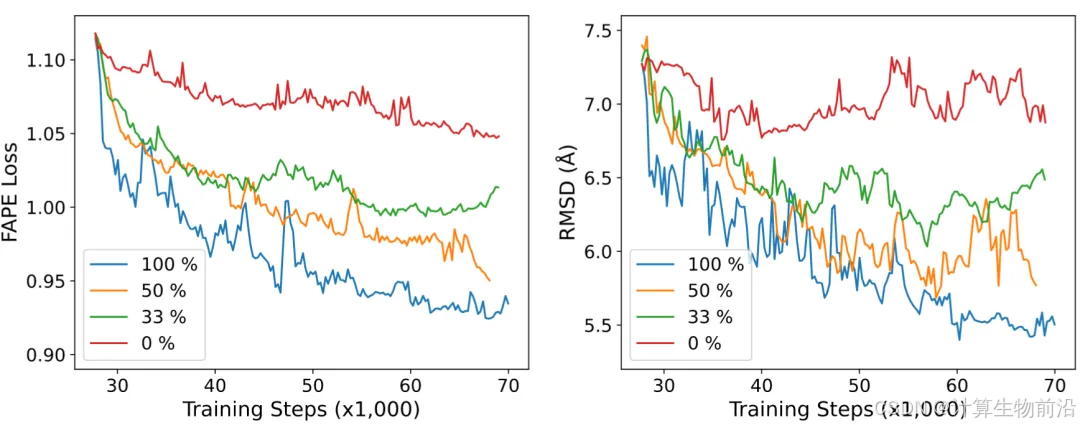
自蒸馏数据的必要性
训练中融合自蒸馏数据(占比75%)显著降低损失函数,提升模型泛化性。
局部几何的精准建模
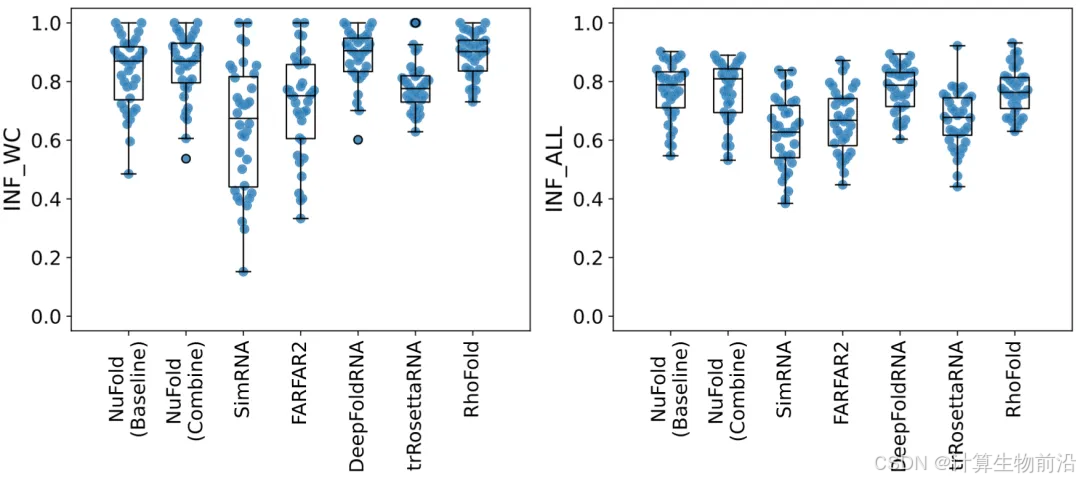
NuFold的灵活核碱基表示精准复现核糖环的C3'-endo和C2'-endo构象(RMSD仅0.03–0.04 Å)。其INF_WC(碱基配对)和INF_stack(碱基堆积)评分超越现有方法,反映更真实的局部作用。
模型具有多聚体预测潜力
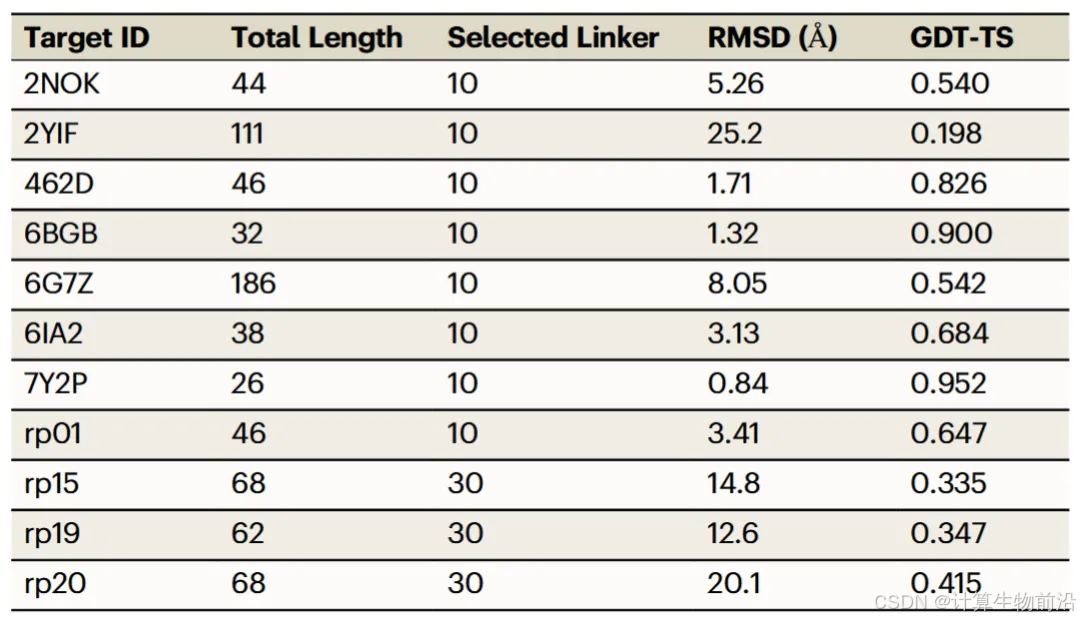
通过序列拼接法(Linker Trick),NuFold成功预测6/11 RNA二聚体结构,如HIV-1 RNA二聚体RMSD达1.71 Å。
案例验证
NuFold对tRNA与核糖开关进行精准建模,尤其在核糖开关的配体结合口袋区域(图b),碱基排列近乎完美。
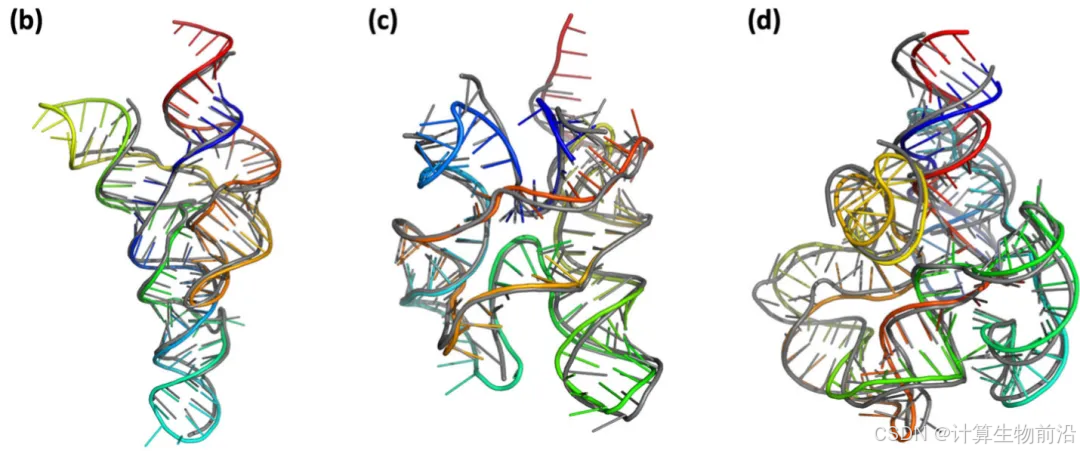
NuFold攻克RNA-Puzzles历史难题,首次实现原子级预测(RMSD 3.55 Å和3.10 Å,图c-d),整体拓扑与实验一致,仅柔性末端或环区存在微小偏差,证明其对复杂全局折叠的解析能力。
参考资料
Kagaya, Y., Zhang, Z., Ibtehaz, N. et al. NuFold: end-to-end approach for RNA tertiary structure prediction with flexible nucleobase center representation. Nat Commun 16, 881 (2025). https://doi.org/10.1038/s41467-025-56261-7

















 1340
1340

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









