







随着计算机技术的发展和计算能力的提升,以模拟和计算的方式去探究实验科学中无法看到的分子机制和细节对目前的科学研究具有极大的促进作用。在分子动力学模拟领域,Gromacs软件表现出独有的优势。本次简要介绍Gromacs的模拟流程和力场构建问题。
1. Gromacs模拟流程
1.1 结构准备
Gromacs的结构输入文件主要为pdb和gro格式。以pdb格式为例,首先要确认结构文件中只包含想要模拟的分析。对于结晶水可用grep –v HOH命令清除,然后通过gmx pdb2gmx命令选择的蛋白N、C端电荷状态,水溶剂模型以及时候忽略原有结构中的H原子,最重要的是通过软件自带的力场文件在该处产生适合体系的力场。
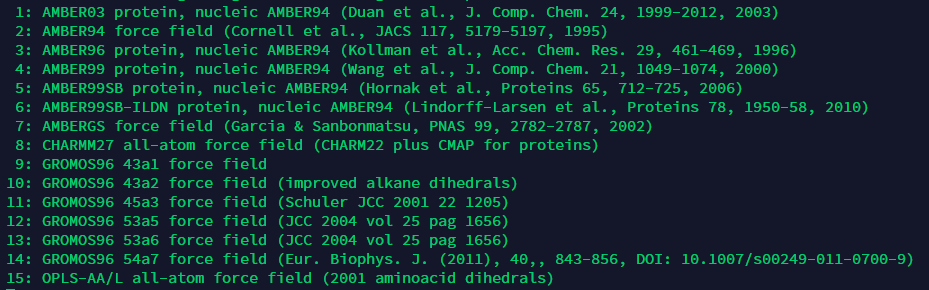
1.2 top文件
pdb2gmx命令完成后会产生一个top文件,文件中包含了力场文件(.itp)和分子数目

1.3 模拟盒子的构建和溶剂填充
使用gmx editconf构建合适尺寸的盒子,利用-bt命令选择盒子的类型。然后利用gmx genbox填充溶剂,同时溶剂分子的数目也会更新到top文件中
1.4 添加离子
首先要ions.mdp文件,利用gmx grompp生成ions.tpr。然后利用gmx genion命令和前面生成的ions.tpr文件生成新的结构文件,同时利用-pname、–nname、-np –nn等指定离子名称和数目,并且利用-p topol.top将添加的离子自动更新到top文件中.

1.5 能量最小化和模拟运行
首先,需要获得能量最小化的mdp文件,利用gmx grompp命令将mdp文件、gro结构文件和top文件打包生成tpr文件。然后,利用gmx mdrun命令让能量最小化模拟运行。
在结果产生的模拟运行中,需要时间和设定更为明确的mdp文件以及能量最小化时产生的结构文件产生tpr,利用gmx mdrun命令提交后续的计算。
2. 力场构建
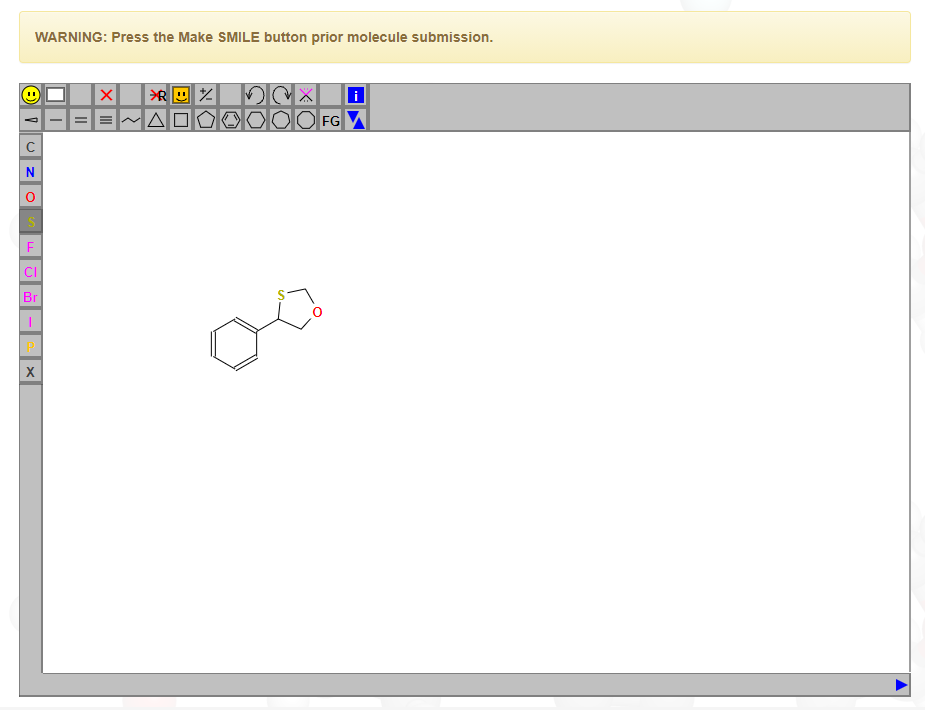
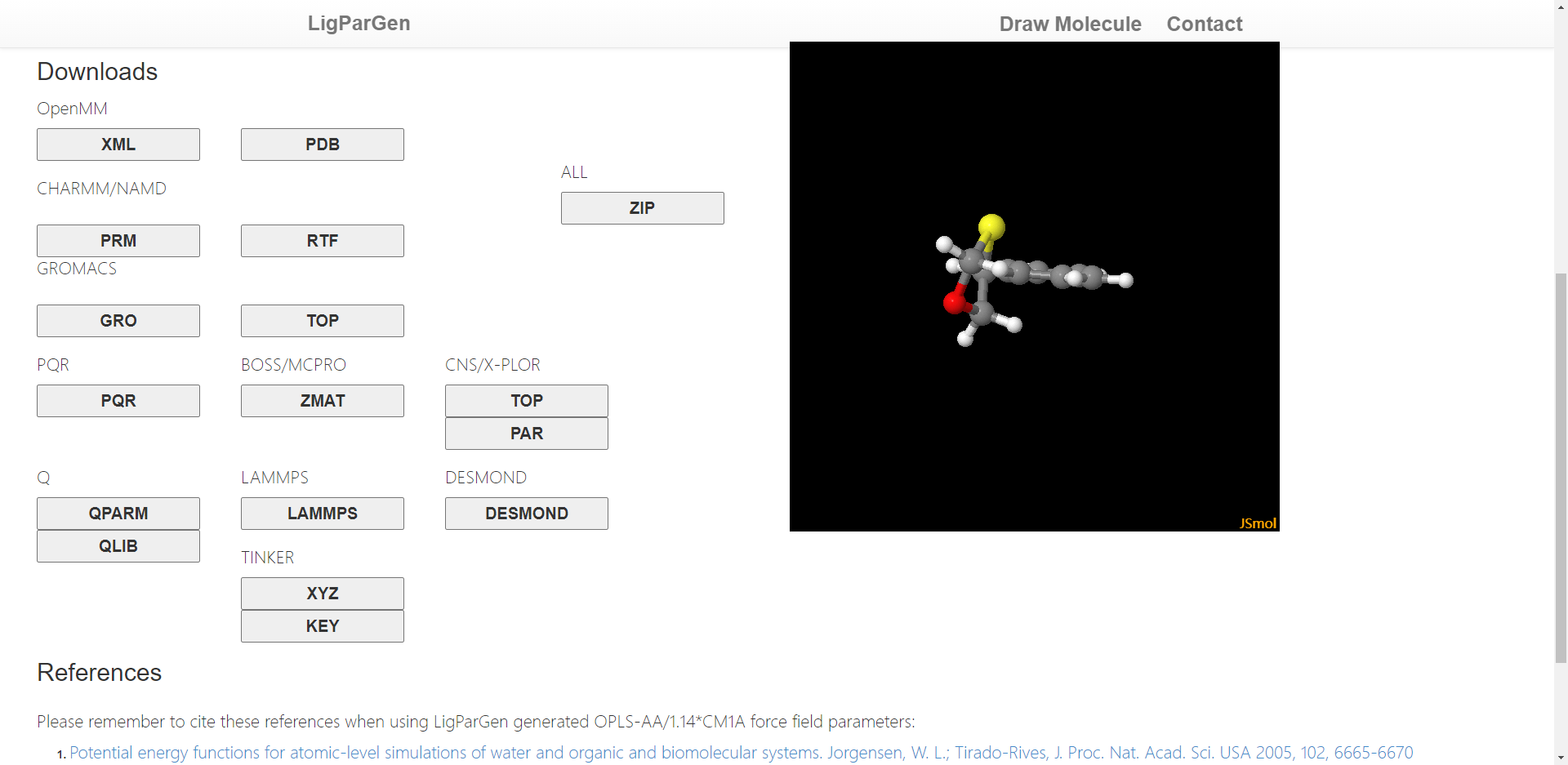
在整个模拟工作的前期,最为关键的事情就是得到准确的力场参数和文件。对于蛋白质等生物分子,Gromacs可以自动识别氨基酸并产生准确的力场文件,但是对于某些特殊的小分子,Gromacs并不能识别和产生力场参数。这时候需要自己构建合适的力场或者利用现有的网站去生成。在此推荐一个力场生成网站:http://zarbi.chem.yale.edu/ligpargen/index.html。该网站需要认为画出小分子的结构,然后会自动识别并产生分子的OPLS全原子力场。
结语
本文简要介绍了Gromacs的操作流程和力场产生的方式。在实际的操作过程中,用户需要准确了解每个命令中的用途以及如何针对自己的需要选择合适的力场类型以及mdp文件中的设定例如计算时长,输出频率,温度和压力的耦合方式等。另外,力场的构建是模拟准备工作中非常关键的一步。由于Gromacs可以识别的分子有限,对于许多工作来说,分子力场的构建非常棘手。本文仅以opls力场为例,用现有的网站辅助生成。实际工作中针对不同的需求,在力场构建时还会需要到一定的文献调研和编程能力。
最后,有相关需求和疑问,欢迎通过微信公众号联系我们。



















 586
586

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











