







引言
未安装好Gromacs的友友,请移步到“《windows系统如何安装Groamcs 怎么选择》”
安装好的友友,对于初学者,下面是一些非常有必要了解的一下Groamcs初学者知识。
Gromacs中所有的文件格式和数据文件解析:《全方位解读GROMACS文件类型:如何快速掌握分子模拟中的关键数据格式和实用技巧 GROMACS文件类型深度解析》
我们假设读者熟悉分子动力学和 Unix,包括使用文本编辑器,例如jot、emacs 或vi。我们还假设 GROMACS 软件已正确安装在您的系统上。当您看到类似这样的行时
ls -l
你应该在你的计算机终端上输入该行的内容。
一、设置你的环境
为了检查您是否有权访问 GROMACS,请首先输入以下命令:
gmx -version
此命令应打印出有关已安装的 GROMACS 版本的信息。里面会包含是否安装了GPU加速版的信息等等。相反,如果此命令返回短语
gmx: command not found.
然后您必须找到您的 GROMACS 版本的安装位置。默认情况下,二进制文件位于初始的安装包目录 ,但是,您可以询问本地系统管理员以获取更多信息,或者找我们专业的远程安装技术进行远程安装 。
二、标题典型模拟流程图
这是在水箱中对蛋白质进行典型 GROMACS MD 运行的流程图。
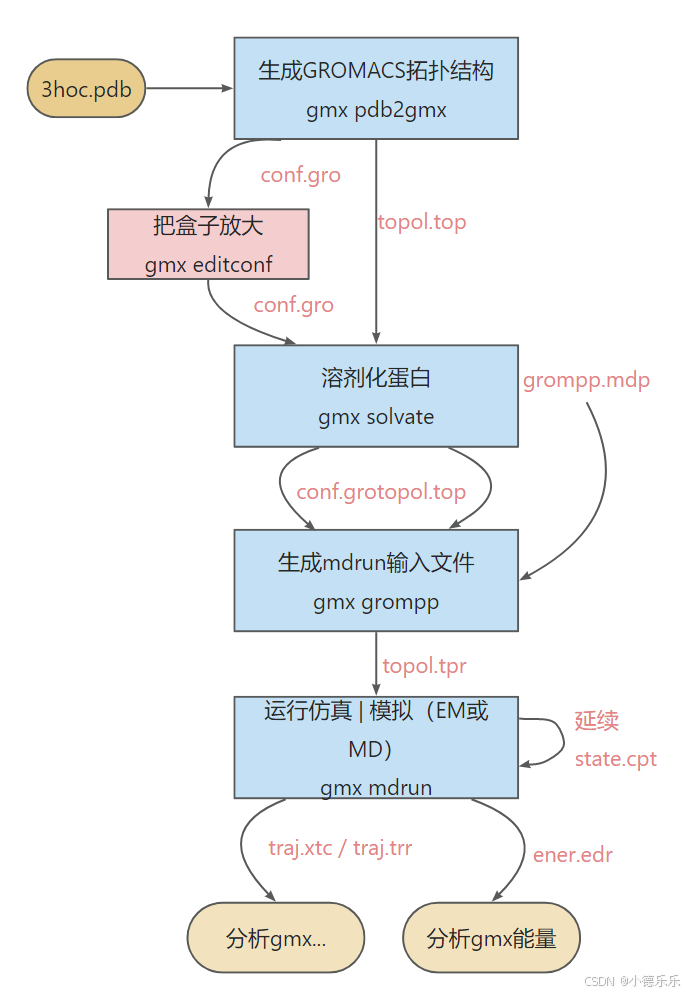
三、重要文件:
以下是您将遇到的最重要的 GROMACS 文件类型的概述。
分子拓扑文件 ( .top)
分子拓扑文件由程序gmx pdb2gmx生成。gmx pdb2gmx将任何肽或蛋白质的pdb结构文件转换为分子拓扑文件。此拓扑文件包含肽或蛋白质中所有相互作用的完整描述。
gmx pdb2gmx的核心呢,可以查看这篇文章《深度解析:如何用GROMACS pdb2gmx 快速生成精准拓扑文件? GROMACS pdb2gmx 使用全解析:从PDB文件到拓扑文件的快速指南》
拓扑#include 文件机制
在顶级文件中构建系统拓扑以呈现给 grompp 时,GROMACS 使用内置版本的所谓 C 预处理器 cpp(在 GROMACS 3 中,它实际上是 cpp)。cpp 解释如下行:
#include "ions.itp"
通过在当前目录、GMXLIB 环境变量指示的 GROMACS 共享/顶级目录以及mdp文件中的-I包含值中的标志指示的任何目录中查找指示的文件。它会找到此文件或报告警告。(请注意,当您提供目录名时,应使用 Unix 样式的正斜杠“/”,而不是 Windows 样式的反斜杠“”作为分隔符。)找到后,它会使用内容,就像您自己将包含的文件剪切并粘贴到主文件中一样。请注意,您不应该自己进行复制和粘贴,因为包含文件机制的主要目的是重复使用以前的工作,使将来的更改更容易,并防止拼写错误。run parameter
进一步cpp解释如下代码:
#ifdef POSRES_WATER
; Position restraint for each water oxygen
[ position_restraints ]
; i funct fcx fcy fcz
1 1 1000 1000 1000
#endif
通过测试预处理器变量是否POSRES_WATER在某处定义(即“如果定义”)。这可以在top文件(或其文件)中较早地使用,在运行参数中使用标志(如上所示),或者在命令行中执行。标志的功能借用了中的类似用法。后面的字符串必须完全匹配;使用不会触发或。此机制允许您更改mdp文件以选择是否要在溶剂而不是top文件上进行位置限制。请注意,预处理器变量与 shell 环境变量不同。
#define POSRES_WATER#include-Dincludecpp-Dcpp-D-DPOSRES#ifdef POSRE#ifdef DPOSRES
分子结构文件 ( .gro, .pdb)
当执行gmx pdb2gmx生成分子拓扑时,它还会将结构文件(pdb文件)转换为 GROMOS 结构文件(gro文件)。pdb文件和 gromos 文件之间的主要区别在于它们的格式,并且gro文件还可以保存速度。但是,如果您不需要速度,也可以在所有程序中使用pdb文件。为了在肽周围生成一盒溶剂分子,使用程序gmx solvate。首先,应使用程序gmx editconf在分子周围定义一个适当大小的盒子。gmx solvate将溶质分子(肽)溶剂化到任何溶剂(在本例中为水)中。gmx solvate的输出是肽在水中溶剂化的 gromos 结构文件。gmx solvate还会更改分子拓扑文件(由gmx pdb2gmx生成)以将溶剂添加到拓扑中。
----> 《解锁高效模拟:利用GROMACS中的gmx solvate工具精准构建和优化分子模拟环境的终极指南 分子动力学模拟小知识 gmx 溶剂化物 核心指令 gmx solvate解析》
分子动力学参数文件 ( .mdp)
分子动力学参数 ( mdp ) 文件包含有关分子动力学模拟本身的所有信息,例如时间步长、步数、温度、压力等。处理此类文件的最简单方法是改编示例mdp文件。示例 mdp 文件 可用。
索引文件(.ndx)
有时您可能需要索引文件来指定对原子组的操作(例如温度耦合、加速、冻结)。通常默认索引组就足够了,因此对于此演示,我们不会考虑使用索引文件。
运行输入文件( .tpr)
下一步是将分子结构(gro文件)、拓扑(top文件)MD 参数(mdp文件)和(可选)索引文件(ndx)结合起来,生成运行输入文件(tpr扩展名)。此文件包含使用 GROMACS 启动模拟所需的所有信息。gmx grompp程序处理所有输入文件并生成运行输入tpr文件。
轨迹文件(.trr,.tng或.xtc)
一旦运行输入文件可用,我们就可以开始模拟。启动模拟的程序名为gmx mdrun 。 通常启动运行所需的gmx mdrun的唯一输入文件是运行输入文件( tpr文件)。gmx mdrun的典型输出文件是轨迹文件(trr文件)、日志文件(log文件)以及可能的检查点文件(cpt文件)。
后续还将更新一些常用的基础教程:
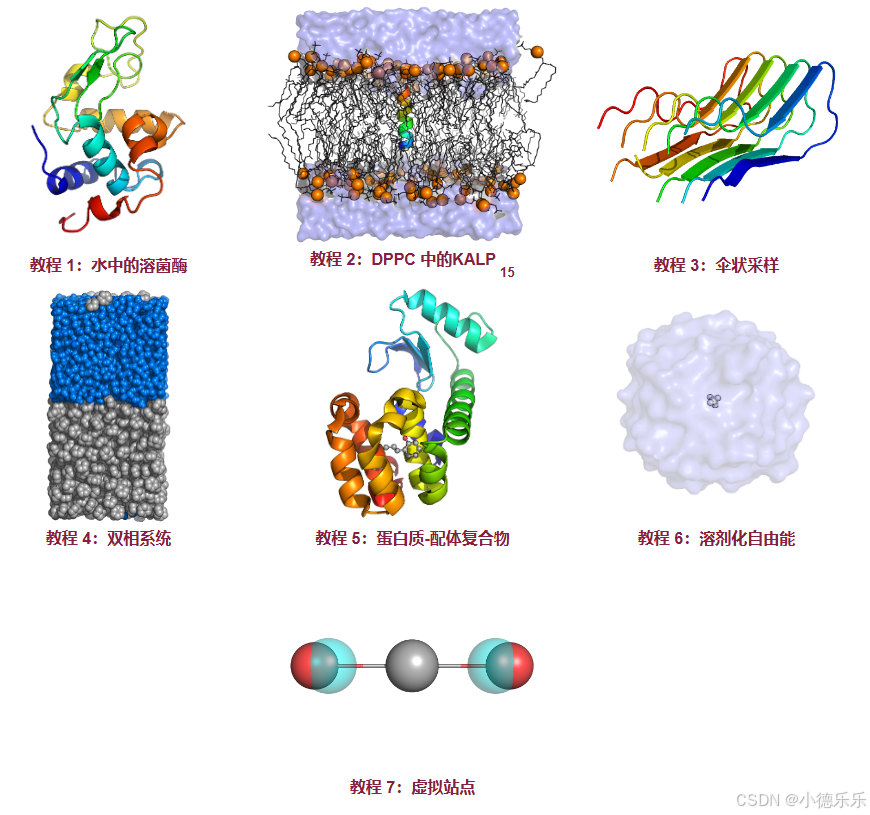
四、后续七个教程:
1. 水中的溶菌酶:本教程旨在向新用户提供使用 GROMACS 在“典型”系统上准备、运行和执行简单分析的工具的基本介绍。
2. DPPC中的KALP 15 :本教程更为高级,专为想要模拟膜蛋白并了解力场结构和修改的更有经验的用户而设计。
3. 伞状采样:本教程也有些高级,适用于希望学习使用伞状采样来计算沿单一线性自由度的平均力 (PMF) 潜力的用户。
4. 双相系统:双相环己烷-水系统的构建。 蛋白质-配体复合物:第五个教程指导用户如何处理蛋白质-配体系统,重点是正确的配体参数化和拓扑处理。
5. 溶剂化自由能:本教程描述了进行简单自由能计算的过程,即消除简单分子(甲烷)和水之间的范德华相互作用。讨论了更复杂的系统。
6. 虚拟位点:本教程指导用户手动构建一个非常简单的线性三原子分子(CO 2)的虚拟位点。
所有这些教程都假设您使用的是 GROMACS 2018 版或更新版本。如果您使用的是旧版本,则并非这里详述的所有功能都可以使用!一些 .mdp 选项和命令行参数在不同版本之间会有所不同,尤其是版本 5.0 和 5.1 中引入的新功能,甚至自 2016.x 系列以来的一些变化。
五、注意事项
官网并不支持类似gmx2020.4_win64_cuda10.2 这样的windows预编译的版本(无团队维护,最高版本也就是2020左右,模拟效果类似,但是模拟时间和分析结果差距过大),推荐大家使用官网源码编译的版本(目前最新版本维护到Gromacs 2024.4 于 2024 年 10 月 31 日发布)。
六、与我联系——远程安装 (不同版本不同价格),平台保证,无需担心。
与我联系——解决AutoDock对接报错以及闪退,提供对接教学服务。
PC端电脑通过
点击PC端分子对接软件合集——“能看到某宝对应的分子对接软件商品!!!。
手机淘宝通过:
点击手淘分子对接软件合集 “——能看到某宝对应的分子对接软件商品!!!

或者直接关注私聊即可。


















 1156
1156

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









