










Whole-genome and genome-wide association studies improve key agricultural traits of safflower for industrial and medicinal use
全基因组和全基因组关联研究改善了用于工业和药用的红花的关键农业性状,红花基因组~

摘要
红花(Carthamus tinctorius)因其种子和花朵而在全球广泛种植。种子中的亚油酸(LA)和花中的水溶性红花黄A(HSYA)是红花可用于工业和药用的关键性状。了解这些性状的遗传控制对于优化红花的品质及其育种至关重要。为了推进这项研究,我们呈现了一种红花品种“川红花1”的染色体级基因组组装,该组装是通过整合Illumina、Oxford Nanopore和Hi-C测序技术实现的。我们获得了1.17 Gb的组装,其contig N50为1.08 Mb,并将所有组装序列分配到12个拟染色体上。红花的进化涉及到了真双子叶植物γ-三倍体事件和一个全基因组复制事件,这导致了大规模基因组重排。自双子叶植物祖先分化以来,发生了广泛的基因组重组。我们进行了代谢物和转录组剖析,并筛选出对种子脂质生物合成有重要贡献的候选基因。我们还分析了参与LA和HSYA生物合成的关键基因家族。此外,我们重新测序了220个红花品系,并使用高质量的SNP数据对八个农艺性状进行了全基因组关联研究。我们识别了与红花中重要性状相关的SNPs。此外,候选基因HH_034464(CtCGT1)被证实参与HSYA的生物合成。总体而言,我们提供了一个高质量的参考基因组,并阐明了红花中LA和HSYA生物合成的遗传基础。这大量的数据将有利于红花中功能基因挖掘和育种的进一步研究。
引言
红花(Carthamus tinctorius),属于菊科或者合翅花科,是一种已在富饶的新月沃土地区培养了约4000年的二倍体植物[1]。这种一年生植物自花授粉,因其种子和花朵在不同领域的多种用途而在全球范围内广泛种植。富含亚油酸(LA)的红花籽油因其有益特性而被视为优质食用油[2, 3]。红花的花朵在全球范围内广泛用于染料、化妆品和食品添加剂[4, 5]。此外,干燥的红花花朵在中国和东南亚传统医学中传统用于治疗各种疾病[6, 7]。红花含有一种叫做水溶性红花黄A(HSYA)的生物活性化合物[8, 9, 65]。由于其显著的潜力,红花有望成为一种值得关注的经济作物[10, 11]。
红花在亚洲、欧洲、澳大利亚和美洲广泛种植,是一种多功能作物(FAOSTAT)。作为一种耐旱作物,随着全球变暖和地方性干旱状况的持续,红花预计将发挥更重要的作用。在种植红花的不同地区,随着时间的推移,出现了具有独特性状的不同品种。然而,红花的遗传分析受到限制,只有少数类型的分子标记用于评估遗传多样性[12–14]。一种红花品种(‘安徽-1’)已通过第二代和第三代测序技术进行了全新测序[15],为遗传多样性和分析提供了宝贵的基因组资源。然而,单一参考基因组无法捕获该物种的所有基因[16–18]。‘川红花1’是在中国四川栽培的一种古老品种,具有独特的农艺性状,例如生长期较短(图1A)。进行全新测序和深入分析是探索其提供的遗传资源的必要步骤。
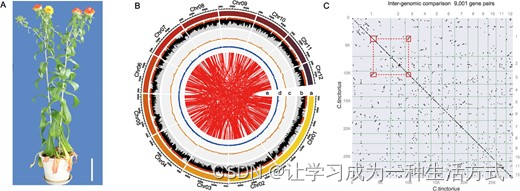
红花基因组装配。A 图为红花“川红花1”照片。标尺=10厘米。B 图中的同心圆代表基因组的不同方面。从最外层到最内层,分别代表染色体长度(a)、重复含量(b)、基因密度(c)、GC含量(d, %)、以及同源块(e)。重复含量超过第三四分位的区域用棕色表示,GC含量高于平均值的区域用蓝色表示。(C)红花基因组同源块的点图。
红花因其油脂含有亚油酸(LA)和花朵含有类黄酮,特别是羟基紫草苷(HSYA)而受到高度评价。这两种成分都具有各种工业和药用价值。油类生物合成的生化过程以甘油三酯(TAGs)的形式在油籽植物中已得到良好建立。它涉及关键酶如二酰基甘油酰基转移酶(DGAT)和脂肪酸去饱和酶(FADs)。DGAT在TAG生物合成中起着关键作用,而FADs将油酸(OA)转化为LA [19-22]。然而,负责红花中LA生物合成的确切基因尚未完全理解。同样,虽然类黄酮的生物合成途径已被广泛记录 [23, 24],但HSYA这一生物活性化合物的特定过程仍不清楚。最近的研究表明,C-葡糖醛酸转移酶(CGT)和P450酶可能参与HSYA的生物合成 [25]。因此,需要进行全基因组范围的筛选,以识别与LA和HSYA生产相关的潜在候选基因。
Table 1
Statistics of the safflower genome.
| Number | Size | |
|---|---|---|
| Contig assembly features | ||
| Sequence number | 3941 | |
| Total bases (bp) | 1 170 951 068 | |
| Minimum sequence length (bp) | 4024 | |
| Maximum sequence length (bp) | 9 019 709 | |
| Average sequence length (bp) | 297 120.29 | |
| N50 (bp) | 1 078 450 | |
| (G + C)s (%) | 38.41 | |
| Chromosome assembly features | ||
| Sequence number | 543 | |
| Total bases (bp) | 1 174 349 068 | |
| Minimum sequence length (bp) | 4024 | |
| Maximum sequence length (bp) | 185 004 703 | |
| Average sequence length (bp) | 18 099 009.73 | |
| N50 (bp) | 96 393 662 | |
| (G + C)s (%) | 38.30 | |
| Ns (%) | 0.29 | |
| Genome annotation | ||
| Gene number | 39 809 | |
| Total gene length (bp) | 213 400 340 | |
| Average gene length (bp) | 5360.61 | |
| Exon number | 235 816 | |
| Total exon length (bp) | 55 574 446 | |
| Average exon length (bp) | 235.669 |
我们采用了一种综合方法,实现了一种古老本土红花品种“川红花1”的染色体级基因组组装。此外,我们对种子和花进行了代谢物分析,以及反映时间和器官特异性变化的转录组剖析,并结合这些分析识别了显著贡献于脂质生物合成的关键基因。我们对参与种子和花中LA和HSYA生物合成的关键基因家族进行了基因组调查。我们重新测序了220个红花品种,并使用高质量的SNPs进行了全基因组关联研究(GWAS),以探索八个关键的农业性状。此外,我们设计实验来证明筛选出的候选基因的功能。这些广泛的数据将对未来功能基因挖掘和工业及医药用途的红花育种研究非常宝贵。
结果
红花基因组特征
我们使用整合方法对“川红花1”红花的基因组进行了测序,结合了Illumina、Oxford Nanopore和Hi-C测序(见补充数据图S1)。Nanopore GridION平台产生了大约111.45 Gb的亚读数,覆盖率约为红花预估基因组大小(大约1.17 Gb,基于Illumina k-mer分析)的100倍(如补充数据图S2和补充数据表S1所示)。过滤后的读数最初被组装并精炼,结果是一个1.17 Gb的组装体,contig N50为1.08 Mb,GC含量为38.41%(详见表1)。经过Hi-C连接后,组装序列被分配到12条染色体(50.84-185.00 Mb)上,N50为96.39 Mb,与细胞遗传学核型分析方法一致(图1B和补充数据图S3)。红花基因组的平均GC含量约为38.30%,不同染色体间的变异很小,其中第10染色体GC含量最高(39.08%),第1染色体最低(36.89%)(补充数据表S2)。我们使用BUSCO方法验证了组装的质量,成功映射率为92.36%,完整基因为97.2%(总数的89.79%),单拷贝映射率为91.42%(总数的84.44%)(补充数据表S3),显示了组装的完整性、连续性和精确性。我们在红花基因组中观察到许多重复的同源块(图1C),这使我们假设红花经历了一次最近的全基因组复制(WGD)事件。
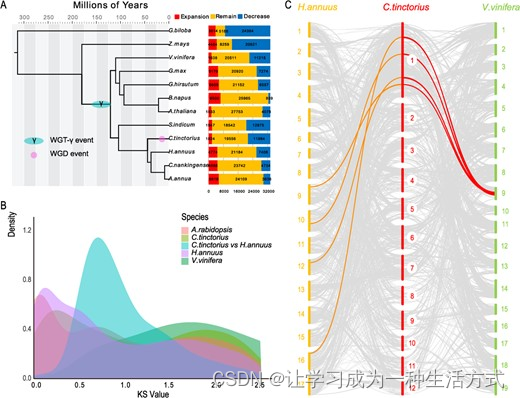
红花的进化历史。A 利用来自12种物种的274个单拷贝基因构建了系统发生树。树中的所有分支的后验概率均超过0.99。该研穊估计了红花全基因组复制(WGD)的时间,树上标记了其他报告的全基因组三倍体(WGT)/WGD事件。柱状图代表基因家族的扩张和缩减。B 分析了红花和其他三种植物物种成对同源副本的同义替换率(Ks)。C 生成了用于比较红花、向日葵(H. annuus)和葡萄(V. vinifera)的同源性图。浅灰线表示同源块。连接线的颜色表示不同物种之间的WGD事件。红色线表示与葡萄相比红花的复制事件更多。橙色线表示红花和向日葵的同时WGD事件。
我们使用自始至终的组合管道和比较方法预测了共39,809个编码蛋白的基因。它们的平均长度约为5.36 kb,平均编码区域为1.39 kb(表1)。超过70%的预测基因使用公共数据库进行了注释,如GO、KO、NR和Pfam等(补充数据表S4)。详细来说,23,251个基因(58.16%)使用GO数据库注释,3,613个基因(9.08%)使用KO数据库注释,14,774个基因(37.11%)使用NR数据库注释,以及23,246个基因(58.39%)使用PFAM数据库注释。重复序列构成了1.17 Gb序列中的约836.16 Mb(71.41%)。有趣的是,这些序列中有39.81%(466.20 Mb)被识别为长末端重复(LTR)元素。LTR元素包括两个主要亚家族,即Ty1/Copia(215.99 Mb,46.33%)和Gypsy/DIRS1(245.91 Mb,52.75%)(补充数据表S5)。
红花基因组的进化
我们将预测的红花基因组与其他11种已测序植物的基因组进行了比较,并聚集了总共33,366个基因家族,包括891个特有于红花的独特基因家族(补充数据图S4)。使用这些物种中保存的274个单拷贝同源基因,我们构建了一个系统发生树,以阐明这些比较物种的进化历史。我们的分析显示,红花(C. tinctorius)和向日葵(Helianthus annuus)大约在4000万年前(MYA)分化,而向日葵和黄花蒿(Artemisia annua或C. nankingense)大约在3200万年前分化(估计),这与分类学分类和化石证据大体一致(图2A)。此外,我们使用CAFÉ来检查红花基因家族的进化和扩张,发现11,984个基因家族经历了收缩,而1,824个基因家族经历了扩张(图2A)。为了调查WGD事件,我们使用了所有基因对在每个染色体片段中检测的同义替换率(Ks)的分布。我们在红花基因组中检测到了两个峰值,分别约为2和0.25,对应于被子植物核心γ三倍体事件和红花特有的WGD事件(图2B)。
为了详细分析,我们选择了向日葵和葡萄的基因组进行植物分子生物学中的同源性映射。基于保守的基因顺序,我们在红花和向日葵之间鉴定了1922个同源块,分别对应于各自基因组中的10,374和13,349个基因(图2C,补充数据图S5)。其中414个块包含超过10个基因。平均而言,红花基因组中的每个块包含五个基因。此外,我们检测到红花和葡萄基因组共有的1135个共线块,分别对应于各自基因组中的9686和8434个基因。平均而言,每个块包含8.5个基因,396个块有超过10个基因。在红花和拟南芥之间,我们鉴定了1170个同源块,分别对应于各自基因组中的6073和6575个基因,平均而言红花基因组中的每个块包含五个基因。在这四种物种中观察到了大规模的基因组重排,如重复、倒置和易位,表明自双子叶植物祖先分化以来发生了广泛的基因组洗牌(图2C,补充数据图S5)。
红花的代谢物、转录组和综合分析
在红花中,油脂含量,特别是LA,和类黄酮,特别是HSYA,是两个关键性状。为了确定种子发育期间的脂质组谱,我们在红花种子的四个不同阶段(命名为SS1、SS2、SS3和SS4)使用UHPLC-MS/MS检测了总脂质代谢物。我们鉴定了218种化合物,将它们分类为13种类型(图3A),其中TAGs包含72种化合物,DAGs(二酰基甘油)包含29种化合物,PCs(磷脂酰胆碱)包含15种化合物,PEs(磷脂酰乙醇胺)包含15种化合物(补充数据表S6)。TAG是主要的脂质(图3B),且脂质含量从SS3阶段到SS4阶段显著增加。我们通过分析不同类型的脂质组成,计算了不同阶段的LA含量。在大部分类型的脂质中,LA的含量超过90%,除了LPC(溶胆磷脂酰胆碱)(21.39%)、PC(70.64%)、SQDG(硫酸根基二酰甘油酮糖醛酸酯)(81.11%)、FA(脂肪酸)(43.96%)和PG(磷酸甘油)(71.54%)。LA含量在SS4阶段最高(图3C)。此外,我们的结果显示,在TAGs中,LA占比最高。我们使用广泛靶向的代谢组方法检测了类黄酮代谢物,鉴定了203种类黄酮代谢物,分为八种类型,包括59种黄酮、41种黄酮C-糖苷、39种黄酮醇、20种黄烷酮、18种花青素、11种异黄酮、2种黄酮木脂素、1种喹诺酮查尔酮和1种生物碱(图3D,补充数据表S7)。
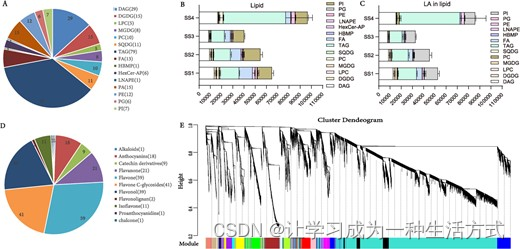
脂质和类黄酮的代谢物及转录组剖析。A 脂质组成及括号内为该成分的数量。B 种子在四个发育阶段的脂质组成。C 各种脂质中的LA含量。D 类黄酮组成及相关数字。E 利用WGCNA对红花的所有转录组数据进行共表达网络分析,包括15,769个基因的20个模块。
为进行基因表达分析,我们对三个不同组别进行了转录组测序:四个发育阶段的花朵、四个发育阶段的种子,以及三种组织类型(根、茎、叶)。过滤掉低质量数据后,我们共获得了197.19 Gb的清洁数据,每个样本的Q20和Q30得分分别高于95%和90%(补充数据表S8)。我们使用Hisat2和StringTie对数据进行了比对和注释,使用红花基因组。组装的片段显示出高度的完整性,映射率从86.52%到92.21%不等,表明测序质量足以进行后续分析。我们利用每百万碱基对每千基因片段数(FPKM)方法(补充数据表S9)分析了表达谱,并观察到不同样本间的显著变异(补充数据表S10)。为进行全面分析,我们使用WGCNA对红花的整个转录组数据进行了共表达网络分析,构建了包含15,769个基因的20个模块(图3E)。
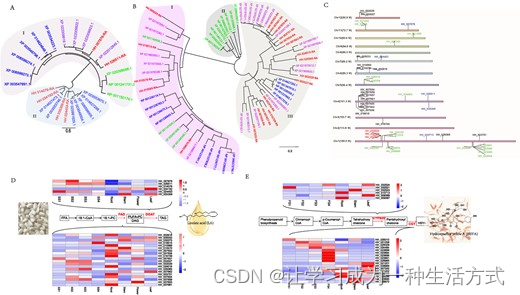
遗传和表达分析,针对LA和HSYA生物合成基因。A DGATs的系统发生分析。I代表DGAT2的亚家族。II代表DGAT1的亚家族。绿色表示来自玉米的DAGTs。蓝色表示来自大豆的DGATs。紫色表示来自向日葵的DGATs。红色表示来自红花的DGATs。B FAD2的系统发生分析。颜色与DGATs的含义相同。C 候选基因的染色体分布。绿色表示CGTs基因,红色表示CYPs,蓝色表示DAGTs,黑色表示FAD2。D DGATs基因表达。E FAD2基因表达。F CYPs基因表达。G CGTs基因表达。
为了进行种子四个不同发育阶段的代谢组和转录组的综合分析,我们使用所有差异基因和脂质建立了O2PLS模型。我们初步判断了不同数据组中相关性和权重高的变量,并筛选出影响脂质的基因(补充数据表S11)。我们的结果显示,HH_016304,注释为过氧化物酶体样酰辅酶A氧化酶3(A),以及HH_015340,注释为脂肪酸ω-羟化酶,这些发现验证了综合分析的准确性。
亚油酸和羟基红花黄甲生物合成的关键基因家族的基因组分析
脂质生物合成过程涉及几个基因家族,其中一个关键酶是DGAT,它作为限制因素。先前的研究表明,不同版本的DGAT有助于饱和和不饱和脂肪酸的生物合成[26]。为了更好地理解DGAT的进化模式,我们特别从红花基因组中提取了属于DGAT家族的单个基因序列,其特征为Pfam域PF03982。通过使用BLAST方法,我们将这些序列与报道的DGAT基因[27]进行比较,并确定了六个同源性,用于构建DGAT家族的系统发生树(补充数据图S6)。发现DGATs分为两个亚家族,即DGAT1和DGAT2。此外,观察到红花中的DGAT2s与向日葵中的DGAT2s有着密切的相似性(图4A)。除了DGAT,另一个广为人知的酶是FAD,它在催化脂肪酸链中形成双键,产生不饱和脂肪酸中起着重要作用。各种类型的FAD酶,如FAD2、FAD3、FAD4、FAD5、FAD6、FAD7和FAD8,已被广泛研究[28]。其中,FAD2被确定为影响油籽植物中三种主要脂肪酸,即油酸、亚油酸和亚麻酸的重要酶[20]。为了研究红花中FAD的进化模式,我们从基因组中提取了所有单一FAD域家族基因序列(PF11960和PF00487),并确定了共20个FAD2基因(补充数据图S7)。系统发生分析显示,FAD2可以分为三个家族,大多数FAD与向日葵中的FAD相关(图4B)。DGAT和FAD基因在基因组中的位置见图4C,大多数FAD基因分布在染色体4上。为了确定哪些DGAT和FAD亚型最可能参与LA生物合成,我们分析了它们的表达。根据我们的结果,HH_026511和HH_024453的DGAT亚型可能在脂质生物合成中起作用,而HH_029365的FAD亚型可能响应种子中的TAG生物合成(图4D)。
HSYA被认为是一个动态元素,是一种含有3,4,5-三羟基环己烷-2,5-二烯-1-酮核心的C-糖苷化合物,2和4位附着β-D-葡萄糖基,6位附着对羟基肉桂酰基。HSYA的生物合成涉及两个基因家族(CYP和UGT),生物合成路径的示意图见补充数据图S8。CYP82D2被认为是具有高底物特异性的F8H,用于黄酮,并且在红花中,F8H与橙皮素查尔酮的4-羟基环己烷相似,因此其类似物可能涉及HSYA的生物合成。我们基于Pfam ID PF00067从红花基因组中提取了403个CYP基因,并根据系统发生树分析在红花中确定了七个CYP82D基因的同源性(补充数据图S9)。红花中的CYP82可以分为三个家族,它们都与向日葵中的CYP82密切相关(补充数据图S10)。参与HSYA生物合成的另一个基因家族是UGT,特别是CGT。已报道CGT参与类黄酮的C-糖基化[29-31]。我们基于PF00201从红花基因组中提取了所有单一UGT家族基因序列,并确定了173个基因,其中19个为推定的CGT基因(1个为CGT,18个为黄酮CGTs)(补充数据图S11)。系统发生分析显示,红花中的UGTs与其他椁物中的UGTs显著不同,表明HSYA路径经历了独立的进化。红花中的CGTs可以分为四个家族,它们都与向日葵中的CGTs密切相关(补充数据图S12)。CYP82和CGTs在红花基因组中的位置见图4C,大部分CGTs分布在染色体1上。为了确定参与HSYA生物合成的候选CYP和CGT,我们分析了它们的表达模式。根据我们的结果,红花花中的HH_032524(CYP)和HH_034464(CGT)具有高表达水平(图4E),表明它们可能参与花中的HSYA生物合成。
通过GWAS分析与红花关键性状相关的基因位点的显著SNPs
我们对220个红花系进行了重新测序,获得了总共7,402,693个高质量SNPs。这些SNPs在12条染色体上的分布在图5A中以视觉方式呈现。使用这些SNPs,我们进行了PCA分析(补充数据图S13)并构建了包含所有系的系统发生树。为了分析群体结构,我们使用ADMIXTURE软件进行了不同K值的分析。交叉验证错误分析表明K=4提供了最优结果(补充数据图S14)。值得注意的是,群体结构分析揭示了所有样本之间没有明显的家族分化,因此表明它们适合进行后续GWAS分析。此外,系统发生树分析揭示了选定群体内四个不同的亚群(图5B),从而确认了早先得出的结论,即K=4确实是通过群体结构分析获得的最佳结果。
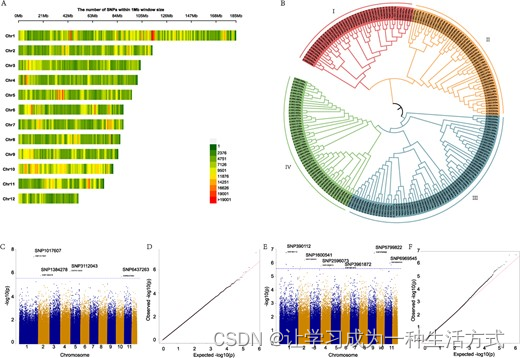
SNP和GWAS分析,与油含量和花色相关。A 红花12个拟染色体中的SNP分布。B 220个红花品系的系统发生树。它可以分为四个亚组。C 与油含量相关的形态特征评估的曼哈顿图。D 油含量的qq图。E 与花色相关的形态特征评估的曼哈顿图。F 花色的qq图。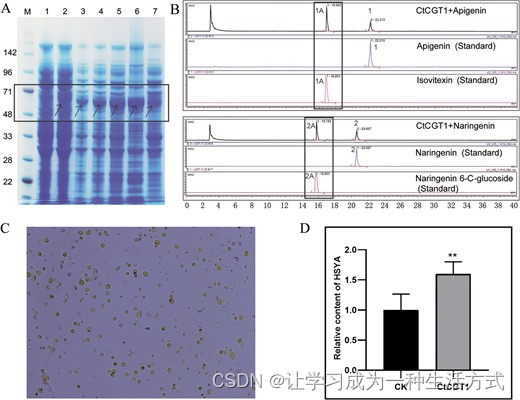
HH_034464 (CtCGT1) 的功能分析。A CtCGT1的原核表达。M是蛋白质标记。通道1,转移CtCGT1未经IPTG诱导后的总蛋白;通道2,转移CtCGT1经IPTG诱导后的总蛋白;通道3-5,经IPTG诱导3、9和16小时后的总蛋白;通道6,使用IPTG培养16小时的细菌经超声破碎后的沉淀物;通道7,使用IPTG培养16小时的细菌经超声破碎后的上清液。箭头指向诱导的蛋百。B 使用粗酶分析CtCGT1的功能。我们使用黄蜀葵素和橙皮素作为底物。作为对照的异维生素和橙皮素6-C糖苷。CtCGT1+黄蜀葵素和CtCGT1+橙皮素表示将黄蜀葵素和橙皮素加入粗酶中。1是黄蜀葵素,1A是异维生素,2是橙皮素,2A是橙皮素6-C糖苷。C 转化CtCGT1的原生质体。标尺=30微米。D 测定HSYA的相对含量。CK是转移空载体后测得的HSYA含量。CtCGT1是转移UBI::CtCGT1后测得的HSYA含量。我们将CK中的HSYA含量默认为1。
我们使用高质量的SNP对八种农艺性状进行了GWAS分析(BN,球花数;BH,分枝高度;BS,苞片刺;FBN,第一分枝数;FC,花色;OC,油含量;PH,植株高度;SD,茎直径)。所有性状数据记录可在补充数据表S12中找到。为确保分析的准确性,使用PLINK软件去除了连锁的SNP并消除了统计模型中连锁位点的干扰。这导致了1,239,895个有效SNP位点的生成。为了减少冗余SNP的数量,我们使用GEC软件基于连锁不平衡准确估计了独立测试的数量(Ne)。总共分析了约1.23亿个SNP标记,并且在红花的所有八种性状中发现了与性状显著相关的SNP。在这些与性状显著相关的SNP中,分别有4个和12个SNP与OC和FC性状显著相关(图5C–F)。与OC相关的4个SNP标记分布在四个不同的染色体上,而与FC相关的12个SNP标记分布在六个不同的染色体上。与FC相关的SNP标记中,HH_018127被注释为YABBY转录因子CDM51。YABBY蛋白是一组参与花发育的植物特异性转录因子[32, 33]。此外,HH_035307是cytochrome_CBB3的成员,这两个基因可能参与花中的类黄酮生物合成。关于与OC相关的SNP,两个SNP(SNP1017607和SNP6437263)导致红花的油含量增加(>26%油含量)(补充数据表S13)。一个新基因(HH_014546)被识别出来。值得注意的是,基于红花种子发育的代谢物和转录组的整合分析,HH_014546也显示出对脂质生物合成的显著影响。所有相关的SNP标记为进一步研究提供了候选基因。详细信息可以在补充数据表S14中找到。
HH_034464参与红花中羟基红花黄甲的生物合成
我们对筛选出的候选基因进行了功能验证,并特别选择了通过整合组学分析确定的HH_034464(命名为CtCGT1)。为了研究CtCGT1,我们采用了广泛使用的原核表达系统进行糖基转移酶的鉴定[34]。首先,我们使用IPTG诱导了CtCGT1的表达(图6A)。CtCGT1的编码序列区域为1320 bp,预测的CtCGT1大小为48.7 kDa。表达载体上的标签约为10 kDa,表明诱导后的CtCGT1加标签应为约60 kDa。在SDS-PAGE上观察到的诱导蛋白实际大小为71 kDa左右(图6A,通道2-7),这表明CtCGT1成功被诱导。此外,可以在上清液中检测到CtCGT1(图6A,通道7),表明其溶解性。使用粗酶液,我们检测了CtCGT1对黄蜀葵素和橙皮素的活性,其中只出现了一个新产物(图6B)。新产生的成分与黄蜀葵素6-C糖苷(异维生素)和橙皮素6-C糖苷的保留时间相同(图6B)。质谱分析还揭示了CtCGT1和黄蜀葵素产物的MS2与异维生素的相同(补充数据图S16A)。同样,CtCGT1和橙皮素产物的MS2与橙皮素6-C糖苷的相同(补充数据图S16B)。这证实了产物是异维生素和橙皮素6-C糖苷,证实了CtCGT1在类黄酮6-C位的糖基化。
在我们之前的研究[35]中,成功制备并使用了红花原生质体来研究花中的类黄酮生物合成的调控。为此,我们在原生质体中过表达CtCGT1(图6C)并随后测量了HSYA含量。结果显示,含CtCGT1的原生质体的HSYA含量与空载体对照组相比有所增加(图6D)。这些发现表明CtCGT1参与了HSYA的生物合成。
讨论
与红花的多种应用相比,这种作物仍未得到充分的重视。获取完整且高度注释的基因组序列对于识别与重要性状相关的基因功能以及植物育种至关重要。在Bowers等人的先前研究中,生成了红花的草图基因组组装,覆盖了预计1.35 Gb基因组的大约三分之二(8.66亿碱基对)。不幸的是,contig N50大小仅为368 bp,scaffold N50大小为1976 bp,这表明由于测序技术和测序深度有限,基因组信息质量较低[36]。Wu等人后来发布了使用PacBio单分子实时(SMRT)和Hi-C测序技术的红花染色体级组装,用于具有高LA含量的“安徽-1”品种[15]。然而,应该注意的是,单一参考基因组可能无法完全代表物种内的所有基因。在中国四川广泛种植的“川红花1”红花是一种古老品种,具有药用和烹饪用途。研究者进行了其基因组的新生测序,并报告了92.36%的成功测序率,97.2%的映射基因是完整的(总数的89.79%),并且91.42%的完整基因具有单拷贝映射(总数的84.44%)(补充数据表S3)。此外,在与Wu等人[15]发布的“安徽-1”基因组比较时,我们的基因组在某些指标上表现更为优越;例如,我们注释的基因数量超过了已发布的基因组,分别为39,809和33,343。这些发现展示了最终序列组装的完整性、连续性和精确性。
在这项研究中,我们分析了脂质和类黄酮的代谢特性。通常使用GC-MS来检测脂质化合物,特别是识别不同类型的脂肪酸[37, 38]。然而,它不能代表原始的脂质组成。在这里,我们使用UHPLC-MS来检测脂质组学。通过分析不同类型的脂质,我们计算了种子发育过程中的LA含量。大多数脂质含量超过90%的LA,亚油酸在SS4阶段显示出最高增长(图4C)。此外,来自TAG的LA百分比最高,PC占LA的大多数(70.64%)。先前的报告表明不饱和脂肪酸通过PC池和PC途径产生[39]。因此,我们的脂质化合物分析结果暗示红花种子中的LA也可能来源于PC池。
在红花种子中,油的主要成分是PUFA,LA约占总油量的76%,α-LA占约0.04%,油中没有γ-LA[40]。FAB2、FAD2和FAD3是参与LA和OA生物合成的关键去饱和酶。FAD2在OA转化为LA中起关键作用,而FAD3通过去饱和LA(18:2)调节亚麻酸(18:3)的含量[20]。FAD2在先前的研究中已被分析[15],但值得注意的是,DGAT在Kennedy途径中控制碳通量向TAG生物合成的过程中也扮演着重要角色,作为速率限制和关键酶[22]。分析DGAT的基因组将提供有关红花种子油中高LA含量的洞察。在植物中,DGAT1和DGAT2是两种主要的DGAT形式,它们在TAG生物合成中具有不同的功能。先前的研究表明,DGAT1在油籽和果实的TAG产生中扮演主要角色[41],而DGAT2在通过积累不寻常脂肪酸的TAG生物合成中至关重要[26]。至于DGAT的表达,HH_026511可能参与红花种子中的脂质生物合成。进一步的研究可以基于这些遗传信息验证这些基因在HSYA生物合成中的功能。
目前,对基本类黄酮生物合成途径已有全面了解[23, 24]。然而,尚未报道任何已知参与HSYA及其衍生物无水红花黄B生物合成的基因,这些都是红花中特有的化合物。考虑到HSYA的结构,有可能两个基因家族CYPs和UGTs在生物合成途径中起作用。其他植物中已报道了几种UGTs,包括柑橘植物中的C-葡萄糖基类黄酮[29],甘草[42]和苜蓿[43]中的O-葡萄糖基类黄酮。在我们的研究中,我们从CGT708基因家族中鉴定了28个假定的CGTs。我们的发现表明,一个基因与查尔酮CGT密切相关,而其余27个基因与黄酮CGT相关。与查尔酮CGT相关的基因可能负责3′ CGT四羟基查尔酮的生物合成。同样,其他27个基因中的一个可能有助于5′ CGT四羟基查尔酮的生物合成。有趣的是,原核表达和原生质体实验的结果表明,HH_034464(CtCGT1)(筛选出的27个基因之一)属于6-糖基转移酶基因家族。这验证了我们筛选结果的准确性。
传统遗传方法的挑战促使了基因组资源的发展,以解决植物栽培相关问题并加速发现重要性状基因。GWAS为识别控制关键性状的位点提供了一个吸引人的选择,并已被广泛用于在经济上重要的植物中识别特定性状的候选基因[44–46],SNP标记有助于分子植物育种。高质量的基因组组装在GWAS中的作图中起着至关重要的作用。在我们的研究中,我们使用综合策略建立了红花的高质量染色体级参考基因组。此外,我们首次收集了来自不同全球遗传背景的220个红芶品系,这对于获得GWAS分析的可靠结果至关重要。这些发展为红花的GWAS分析提供了优秀的基础。
除了与OC和FC性状分别显著相关的4个和12个SNPs外,我们还鉴定了与BH性状相关的5个SNPs,与BN性状相关的42个SNPs,与BS性状相关的104个SNPs,与FBN性状相关的51个SNPs,与PH性状相关的84个SNPs,以及与SD性状相关的17个SNPs(补充数据图S15和补充数据表S14)。仍有一些候选基因需要进一步验证。尽管如此,所有结果都提供了有价值的功能基因分析数据,这将有助于推动红花的工业和药用育种。
材料与方法
材料
我们采用红花品种“川红花1”进行基因组测序。这个品种在成都中医药大学温江校区的药用植物园中种植。我们的实验室通过自交繁殖维持了这个品种超过10年。为了检测类黄酮,我们使用了授粉后约3天的盛开花朵。对于脂质分析,我们检查了不同发育阶段的种子。为了分析表达谱,我们在授粉后3天收集了根、茎、叶和花,以及四个不同发育阶段的种子。为了提取总RNA,我们遵循了制造商的说明,并使用了Trizol(Invitrogen, CA, USA)。通过与合作单位的合作,我们从世界各地获得了220个红花品系,每个品系都经过了自交并进行了表征。在2020年,我们评估了这些红花品系的农艺性状。
基因组测序、组装和注释
我们采用CTAB方法从红花叶片中提取基因组DNA。为了创建纳米孔图书馆,我们产生了10 kb片段,并使用Nanopore GridION系统获得了平均长度为3.75 kb的29.71 Mb纳米孔读数。此外,我们还使用Illumina HiSeq X Ten仪器生成了配对末端Illumina文库。对于Hi-C测序,我们使用新鲜采摘的红花叶片。所得产品经过富集,物理剪切至350 bp,并进行了DNA纯化。从未连接的末端去除生物素。为了估计红花的基因组大小,我们使用带有jellyfish工具(−m 21 -s 1000000000)v2.3.0 [47]的Illumina clean reads。使用Canu (corOutCoverage = 80) v1.71 [48]生成初步的纳米孔读数,然后使用NextPolish v1.3.1 [49]利用Illumina clean reads改进了contigs。使用LACHESIS v1根据Hi-C数据连接contigs,采用默认参数。重组的contigs通过使用Create Scaffolded Fasta.pl进行了完善。基因预测使用EvidenceModeler v1.1.1进行,该工具结合了ab initio基因预测工具,如Augustus v3.3.1、Gene Mark-ES v4.3.3和SNAP v20131129,以及同源基因预测工具如BLAST v2.2.28和GeneWise v2.2.0。同源基因预测使用的参考资料是拟南芥数据。为了生成和训练基因模型,我们使用PacBio的完整ISO-seq管道,并通过Trinity v2.8.5 [50]生成RNA-seq组装。使用BUSCO v3.0.0 [51]评估了组装和基因预测。使用RepeatMasker v4.0.7进行重复元素的注释。此外,我们使用RepeatModeler v1.0.8 [52]构建了一个de novo数据库,以REPBASE作为比较的参考。
基因组进化分析和构建系统发生树
我们使用Hcluster_sg v0.5.1对单拷贝同源基因进行分组,随后使用MUSCLE v3.4进行序列比对。使用PhyML v3.0 9 [53]构建最大似然系统发生树,并使用R8s包v1.81推断分化时间。校准时间从TimeTree网站(TimeTree :: The Timescale of Life)获取。使用CAFÉ v4.2.1分析基因家族的扩张/收缩,并使用MCscan v0.9.6 [54]进行同源性比较,采用默认参数。
脂质提取和UHPLC–MS/MS分析
我们根据之前的研究[65]采用了一种脂质提取技术。测量大约30 mg的样本,并加入100 μl的全脂质内标准物(10 μg/ml)。混合物在-20°C下与2 ml的甲醇激烈搅拌12小时,随后加入2 ml的二氯甲烷并搅拌1小时。再加入2 ml的二氯甲烷和1.6 ml的超纯水,然后离心。丢弃上层,将下层与第二次提取的下层合并。然后使用氮气吹干合并物,用1 ml的异丙醇重构,通过有机膜过滤器(孔径0.22 μm)过滤,并进行LC–MS/MS分析。UHPLC–MS/MS评估在中国北京的一个设施进行,使用Vanquish UHPLC系统(Thermo Fisher, Germany)连接到Orbitrap Q Exactive™ HF质谱仪(Thermo Fisher, Germany)。溶剂梯度和质谱仪设置经过优化以确保精确分析。
类黄酮提取和UHPLC–MS/MS分析
我们根据之前的研究[66]采用了一种提取花朵的技术。冻干花朵使用搅拌磨和锆珠粉碎,随后在4°C下使用70%的水性甲醇进行过夜提取。然后,提取物通过SCAA-104滤器过滤,滤器孔径为0.22 μm,然后进行LC–MS分析。使用的UHPLC柱是Waters ACQUITY UHPLC HSS T3 C18,采用梯度程序和优化的溶剂系统进行分析。流出物连接到ESI三重四极杆-线性离子阱(Q TRAP)-MS仪器,使用MRM实验在正离子模式下进行LIT和QQQ扫描。仪器调谐和质量校准使用聚丙二醇溶液在QQQ模式下执行。DP和CE针对每个MRM转换进行优化,同时根据该时期内洗脱的代谢物监测特定的MRM转换集。
代谢物数据分析
使用www.r-project.org的“prcomp”函数进行了无监督PCA分析,这是一个流行的R统计库。在进行无监督PCA之前,为了标准化数据,实施了单位方差缩放。代谢物和样本的层次聚类分析(HCA)结果通过包含树状图的热图展示。使用R中的“cor”函数获得样本间的Pearson相关系数(PCC),并以热图形式表示。HCA和PCC分析均使用R中的热图包进行。在HCA中,代谢物的标准化信号强度(使用单位方差缩放)通过颜色光谱可视化。基于VIP值≥1和绝对log2(倍数变化)≥1选择差异代谢物。VIP值来自OPLS-DA结果,包括使用R包MetaboAnalystR创建的得分图和置换图。在运行OPLS-DA之前,数据应用了Pareto缩放,通过进行200次置换测试解决了过拟合问题。
RNA测序和数据分析
使用Illumina HiSeq平台进行了RNA测序。通过使用Trimmomatic包过滤和修剪,获得了高质量的清洁读数。使用HISAT2将读数对齐到红花基因组,使用StringTie生成转录本及其表达。通过FPKM方法量化转录本丰度。设置折叠变化≥1和调整后的P值≤0.01为显著性测试阈值。进行基因本体(GO)分析,采用功能分类,而通过KEGG检索获得基因和基因组百科全书(KEGG)分类图。
亚油酸和羟基红花黄A生物合成相关基因家族的筛选
根据Pfam注释,在红花基因组中鉴定了参与HSYA生物合成的同源基因,包括CYP82和UGTs,以及参与LA生物合成的基因,如FADs和DGATs。参考了Liu等人[55]研究中的FAD基因家族,Zhao等人[56]研究中的CYP基因家族,Ito等人[29]研究中的CGT基因家族,和Turchetto-Zolet等人[27]研究中的DGAT家族。通过将所有序列提交到Ensembl Plants网站,验证了保守结构域。CYP82基因家族对应于PF00067,而CGT家族对应于PF00201。DGAT家族对应于PF03982,包含在甘油磷脂生物合成中起关键作用的磷脂酰甘油酰转移酶(LPLAT)结构域。FAD家族对应于PF11960和PF00487,包含参与ω-3脂肪酸去饱和酶的PLN02498和另一个响应于ω-6脂肪酸去饱和酶的重要结构域PLN02505。此外,大多数FAD基因具有膜-FADS样结构域,这是一种非血红素、含铁的、依赖氧的酶,参与脂肪酰基脂肪链中双键的区域选择性引入。这些酶在维持生物膜的适当结构和功能中起着重要作用。最后,使用Geneious 11软件中的最大似然法构建了每个基因家族的系统发生树,bootstrap值为1000。
GWAS分析
我们使用GATK 4.0管道对220个红花品系进行了重新测序,以识别突变和过滤SNP位点。具体来说,采用GATK推荐的过滤标准,如Variant Filtration -filter-name FS和-filter 'FS > 30.0',-filter-name DP和-filter 'DP < 5',以及-filter-name MQ和-filter 'MQ <= 50.0'。使用vcftools [58]进行额外的过滤,以实施一组特定标准,包括maf < 0.05,过滤掉缺失率>0.1的位点,并仅保留二等位位点,以确保后续分析的质量。我们使用Beagle [59]对所有保留的位点进行位点错误校正和基因型填充。使用PLINK 1.90 [60]实现PCA,分析了10个主成分并绘制了前两个成分。使用ADMIXTURE软件[61]通过进行从K=2到K=8的1000次迭代,并使用交叉验证误差选择最佳K值,分析了群体结构。使用Treebest软件[62]进行系统发生分析,并使用iTOL软件(iTOL: Interactive Tree Of Life)实现数据可视化。ADMIXTURE分析显示植物材料没有显示出显著的家族分化,这导致我们进行GWAS分析。为了减少连锁位点对统计模型的影响,采用PLINK软件的特定过滤标准-indep-pairwise 100 10 0.2。使用EMMAX软件[61]进行GWAS测试,将八个不同性状(BN,球花数;BH,分枝高度;BS,苞片刺;FBN,第一分枝数;FC,花色;OC,油含量;PH,植株高度;SD,茎直径)与相关矩阵(K模型)相关联。所有性状数据记录可在补充数据表S12中找到。使用GEC软件[62]估计有效独立测试(Ne)以减少SNP信息的冗余,设置P < 2.72e−6(P = 1/Ne,Ne = 367544)为显著关联的阈值。最后,使用ANNOVAR软件[63]对SNP位点进行注释。
重组蛋白表达和酶反应产物检测
将CtCGT1基因插入pET32a载体(Thermo Fisher, USA)中以实现原核表达。所得重组载体称为pET32a-CtCGTs,然后使用热休克方法引入细菌(BL21, DE3)菌株中。在1升的LB培养基中补充50 μg/ml氨苄青霉素,在37°C下以200 rpm的速度摇动培养BL21细胞。细胞生长至OD600达到0.5。通过在冰上冷却烧瓶10分钟后加入120 μM的IPTG诱导细胞。为了确定最佳诱导时间,进行了从3到16小时的一系列梯度实验,最终采用过夜诱导进行后续实验。然后细胞在16°C下以200 rpm培养16小时,收集并重悬细胞沉淀于30 ml裂解缓冲液(25 mM HEPES pH 8, 500 mM NaCl, 5 mM咪唑)中。在冰上进行超声波破碎以裂解细胞,随后在4°C下以6517 g离心15分钟去除上清液。最后,使用所得细胞沉淀进行初步酶活性检测。为了评估CtCGT1的活性,使用原核表达获得的粗酶液。反应产物使用UHPLC结合相应的糖苷标准进行分析。反应条件包括1 mM受体底物、2 mM UDP葡萄糖、10 μM纯化的重组蛋白和50 mM Na2HPO4-NaH2PO4缓冲液(pH 8),反应体积为100 μl。反应在25°C下进行5分钟。随后,迅速加入200 μl预冷的甲醇以实现高效混合和均质化。以12000 rpm离心15分钟,收集上清液以进行进一步分析。对于UHPLC分析,使用Agilent Eclipse Plus C18柱(150 mm × 3.0 mm,1.8 μm),柱温设置为30°C。流动相由含0.1%甲酸的纯水(A)和乙腈(B)组成。采用梯度洗脱过程,开始时为5%相B,在0–40分钟内达到95%相B,流速为0.2 ml/min。


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









