









Quantitative profiling of m6A at single base resolution across the life cycle of rice and Arabidopsis
水稻和拟南芥生命周期中单碱基分辨率的m6A定量分析
“Xian”(籼)和“Geng”(粳)米怎么读?_一个米一个更念什么-CSDN博客

摘要
N6-甲基腺苷(m6A)在调控mRNA代谢中发挥着重要作用。然而,目前尚未报道在不同植物组织中具有单碱基精度的全面m6A甲基化图谱。在本研究中,我们使用m6A-SAC-seq技术构建了水稻和拟南芥不同组织中转录组范围的m6A单碱基分辨率图谱。分析结果显示,水稻中共有205,691个m6A位点分布在22,574个基因上,而拟南芥中有188,282个m6A位点分布在19,984个基因上。保守的m6A位点在水稻和拟南芥的直系同源基因对中参与了组织发育、光合作用以及胁迫响应的调控。我们观察到在某些植物组织中,3’UTR的m6A位点对mRNA具有总体稳定作用。与哺乳动物类似,植物mRNA中(除最后一个外)内含子长度与m6A水平呈正相关。此外,研究数据表明,植物mRNA的停码子附近存在一个活跃的m6A沉积过程。此外,MTA催化的mRNA m6A位点与促进或抑制翻译均有关联,呈现出更复杂的调控模式。因此,本研究提供了与植物功能相关的单碱基分辨率m6A位点的深入资源,并揭示了跨物种控制m6A生成的抑制-激活模型。
引言
RNA 修饰是调控 mRNA 加工和代谢的重要调节因子,包括剪切、3′ 末端加工、核输出、翻译和降解。N6-甲基腺苷(m6A)是哺乳动物和植物中最常见的 mRNA 内部修饰之一。研究表明,在拟南芥中,有两种不同的 m6A 转移酶复合物能够在 mRNA 上安装 m6A。第一种甲基转移酶复合物由哺乳动物 m6A 甲基转移酶复合物的五种直系同源成分组成,包括 mRNA 腺苷甲基酶(MTA)、MTB、VIRILIZER (VIR)、FKBP12 结合蛋白 37KD (FIP37) 和 E3 泛素连接酶 HAKAI。相比之下,FIONA1 (FIO1) 是人类甲基转移酶 METTL16 的拟南芥同源物,也在 U6 小核 RNA 和部分 mRNA 中引入 m6A 修饰。m6A 转移酶复合物中的蛋白缺陷,例如植物中 MTA 的功能丧失,会导致胚胎致死表型以及胁迫响应的异常。此外,研究表明拟南芥中 FIP37 调控茎尖干细胞命运,FIO1 调控开花转变和叶绿素稳态,而 VIRILIZER (VIR) 对维管发育至关重要;而在水稻中,FIP37 的缺陷会导致小孢子的早期退化。
m6A 甲基化是可逆的。拟南芥中的 RNA m6A 去甲基酶 ALKBH10B 和 ALKBH9B(人类 m6A 去甲基酶 ALKBH5 的同源物)影响了开花转变和病毒感染。我们的研究发现,过表达哺乳动物 m6A 去甲基酶 FTO 可显著提高水稻和马铃薯的生物量和产量,表明调控 RNA m6A 甲基化可能是未来改良作物的一种有前景的育种或工程策略。尽管这些研究揭示了植物和动物中保守的 mRNA m6A 调控机制,但植物中高分辨率的 mRNA m6A 图谱仍未绘制出来,m6A 在植物发育和其他途径中的分子作用机制大多尚不清楚。我们使用最新开发的高分辨率测序技术填补了这一空白。
甲基化 RNA m6A 免疫共沉淀测序(MeRIP-seq 或 m6A-seq)已被广泛用于检测动物和植物中富含 m6A 的转录本。然而,这种方法缺乏单碱基分辨率,无法量化修饰程度。虽然包括 m6A 单核苷酸分辨率交联与免疫共沉淀(miCLIP)在内的 MeRIP-seq 变体能够检测单碱基分辨率的 m6A 位点,但这些方法通常表现出较低的紫外线交联效率,且无法评估修饰化学计量比。无抗体的单碱基 m6A 描述方法,如 m6A-REF-seq 或 MAZTER-seq,能够检测 ACA 基序中的 RNA 修饰,但无法识别其他甲基化基序。此外,纳米孔直接 RNA 测序(DRS)已被用于定位 m6A 位点,但在量化 m6A 修饰水平差异方面仍面临挑战。我们最近开发的 m6A 选择性烯丙基化学标记与测序(m6A-SAC-seq)能够以单核苷酸分辨率精确定位全转录组范围内的 m6A 修饰位点。
通过 m6A-SAC-seq,我们绘制了水稻和拟南芥不同组织类型中 mRNA 的单核苷酸分辨率 m6A 图谱,并识别出跨越水稻和拟南芥生命周期的大量高置信度 m6A 位点。保守的 m6A 修饰位点调控组织发育、光合作用和胁迫响应。m6A 水平与内部外显子长度呈正相关,但最后一个外显子例外。跨人类、水稻和拟南芥的 m6A 比较分析揭示了一种特有的 m6A 分布模式,由抑制-激活双重模型控制 m6A 沉积。我们发现,水稻和拟南芥的 mRNA 在 3’UTR 的 m6A 修饰比例高于哺乳动物系统,这些修饰通常稳定 mRNA 并增强翻译,与 m6A 含量呈良好相关性。此外,MTA 在拟南芥核基因组和叶绿体基因组的光合作用相关基因上介导 m6A 沉积,这些 m6A 位点可以以路径依赖的方式促进或抑制翻译效率。总之,我们提供了水稻和拟南芥的高分辨率和定量的 m6A 修饰图谱,为未来研究 m6A 在植物发育和进化中的调控作用以及植物工程的应用提供了重要基础。
结果
m6A-SAC-seq 鉴定了水稻和拟南芥中的 m6A 修饰位点
m6A-SAC-seq 利用二甲基转移酶 MjDim1 在 m6A 上引入烯丙基,该烯丙基在化学诱导的环化反应后,可在逆转录过程中被读取为突变信号(补充图 1a)。我们从拟南芥的九种组织(幼苗、茎尖、根、莲座叶、茎生叶、茎、花、果荚和种子)以及水稻的八种组织(暗处理的胚芽、光处理的胚芽、8 天幼苗、2 周幼苗、花序、开花后 10 天的旗叶、开花后 10 天的胚乳和开花后 10 天的胚)中提取了总 RNA,每种样本有两个生物学重复(图 1a, b)。
从每个生物学重复样本中提取的 polyA 尾 RNA 经过纯化后,采用 LC-MS/MS 测定其 m6A/A 比值。在拟南芥的不同组织中,polyA 尾 RNA 的 m6A/A 比值在 0.36–0.75% 范围内变化(图 1c),而在水稻中,该比值在 0.52–0.67% 范围内变化(图 1d)。剩余的 polyA 尾 RNA 随后按照 m6A-SAC-seq 文库构建协议进行处理,用于单碱基分辨率 m6A 位点的定位。
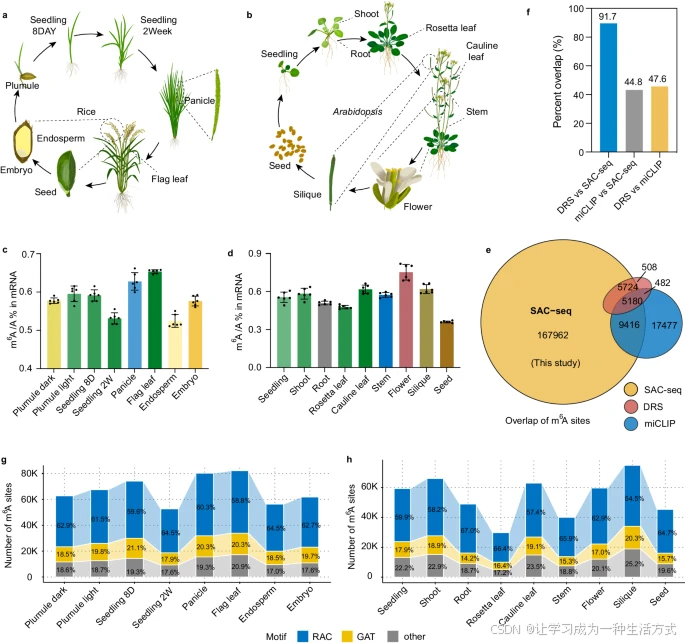
m6A-SAC-seq 鉴定了水稻和拟南芥中的 m6A 修饰位点
a, b. 收集了水稻的八种组织(a)和拟南芥的九种组织(b)贯穿其生命周期,并使用 m6A-SAC-seq 进行分析。 c, d. 使用 LC-MS/MS 测定水稻(c)和拟南芥(d)样本中的 mRNA m6A 水平。通过校准标准计算 m6A/A 的比率,数据以均值 ± 标准差表示(n = 6)。 e. 使用 ±5 nt 滑动窗口比较 m6A-SAC-seq 检测到的 m6A 位点与 miCLIP 和 DRS 方法鉴定的位点之间的重叠情况。 f. 不同方法鉴定的 m6A 位点重叠百分比的比较。 g, h. 水稻(g)和拟南芥(h)组织中 m6A 位点数量及其基序分布,基序分为 RAC、GAT 和其他组别。碱基 "R" 代表 A 或 G。8D 表示 8 天,2W 表示 2 周。数据来源于 Source Data 文件。
为减少背景噪声并消除潜在的批次效应,我们在每个样本中加入了 2% 的 spike-in 校准探针,这些探针包含不同比例的 m6A 修饰 NNm6ANN 基序。通过确定这些探针中 A 和 m6A 的突变率,我们可以最准确地评估每个样本中单个位点的 m6A 修饰比例。初步结果表明,植物细胞中校准探针的标记效率很高,与哺乳动物细胞中观察到的结果相当(补充图 1b)。此外,未修饰的 A 位点的平均背景噪声低至 0.49%,远低于检测 m6A 位点的截止值(<5%),表明从这些植物样本中获得的 m6A-SAC-seq 数据质量较高。
我们进一步评估了每个样本的相对转化率和背景噪声。拟南芥样本中的相对转化率范围为 0.92 至 1.11(补充图 1c),而水稻样本中的范围为 0.96 至 1.07(补充图 1d),显示了样本间 m6A 标记效率的一致性。通过仔细校准每个样本中观察到的差异,我们确保了不同样本间 m6A 水平的公平和准确比较。
在进行下游分析之前,我们将 m6A-SAC-seq 数据与先前发表的通过 DRS 和 miCLIP 方法在拟南芥中分析的 m6A 位点进行了比较。只有在所有组织中具有足够序列覆盖(深度 >10)的位点被选择用于进一步分析。结合本研究中 m6A-SAC-seq 检测到的 m6A 位点后,共获得 188,282 个 m6A 位点,其中约 42% 和 17% 分别与 DRS 和 miCLIP 测得的 m6A 位点重叠(补充图 1e)。在 ±5 nt 滑动窗口内,更多先前检测到的 m6A 位点与 m6A-SAC-seq 位点重叠(图 1e, f)。如预期,与 miCLIP 方法相比,m6A-SAC-seq 与 DRS 检测的 m6A 位点重叠比例更高(图 1f),可能表明 miCLIP 在植物组织中因交联效率低而导致较低的准确性。
主成分分析(PCA)显示,不同组织的 m6A 分数具有显著的聚类(补充图 2a–d)。在拟南芥文库中,平均从 12,652 个基因中鉴定出 49,791 个 m6A 位点;在水稻中,平均从 15,138 个基因中检测到 67,173 个 m6A 位点。水稻八种组织中高置信度 m6A 位点的数量从 2 周幼苗的 52,646 个到旗叶的 82,157 个不等(图 1g),而拟南芥九种组织中的 m6A 位点数量范围为种子的 25,990 个到果荚的 74,259 个(图 1h)。与先前发布的拟南芥幼苗 MeRIP 数据(7,489 个 m6A 峰)相比,我们在幼苗数据集中鉴定了约 59,212 个 m6A 位点,覆盖 14,180 个基因,显示了 SAC-seq 方法的高灵敏度。因此,拟南芥幼苗转录组每个基因平均含有约 4.2 个 m6A 位点,比 MeRIP 数据高出四倍。类似地,水稻组织中每个基因平均含有约 4.4 个 m6A 位点,这表明不同植物物种之间可能存在保守的分布密度。与哺乳动物和植物中先前的发现一致,RAC(R = A 或 G)基序在水稻和拟南芥中均显示出最高的甲基化基序频率(图 1g, h)。
不同组织中的 mRNA m6A 单碱基分辨率图谱
我们进一步分析了拟南芥和水稻全转录组中的 m6A 分布。拟南芥和水稻的所有鉴定的 m6A 位点分别可以通过 GEO 编号 GSE245738 和 GSE243722 访问。
在元基因分布图中,大多数 m6A 位点在拟南芥(图 2a)和水稻(补充图 3a)中高度富集于 3′ 非翻译区(3′ UTR),其次是编码区序列(CDS)和 5′ 非翻译区(5′ UTR)。虽然哺乳动物和植物的 mRNAs 都在 3′ UTR 中高度富集 m6A(图 2b),但水稻和拟南芥的 mRNAs 在 3′ UTR 中的总体 m6A 修饰比例显著高于人类 HeLa 细胞(图 2b)。
除了 3′ UTR 和 CDS 区域中的 m6A 位点外,我们还在水稻和拟南芥的内含子和 5′ UTR 区域中观察到大量的 m6A 位点,这与哺乳动物中观察到的结果一致(补充图 3b, c)。内含子区域的平均 m6A 比例显著高于 5′ UTR 和 CDS,但低于 3′ UTR(补充图 3b, c)。
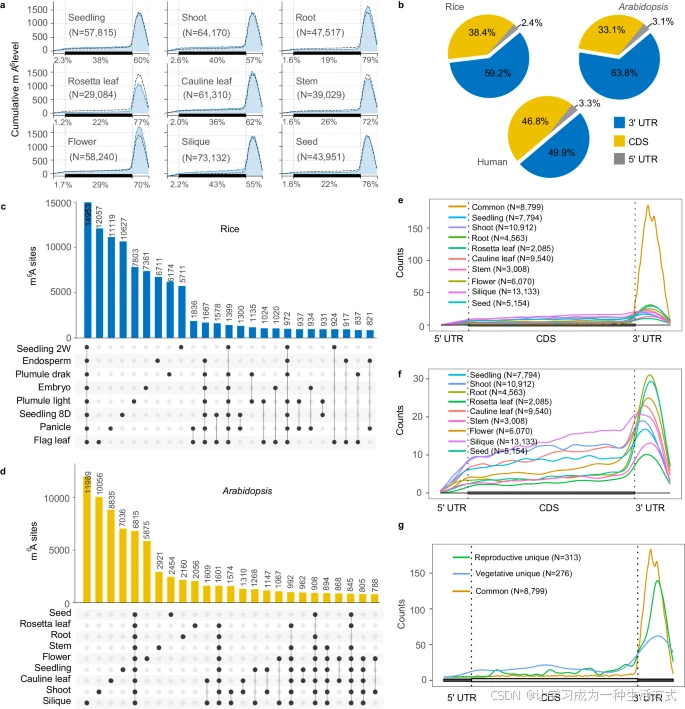
a 和 b 元基因分布图展示了拟南芥九种组织中 m6A 位点在转录本中的分布。每个转录本分为三个区域:5′ UTR、CDS 和 3′ UTR。黑色虚线表示九种组织中平均 m6A 比例。图中标注了 m6A 位点数量(N),以及各组织中 5′ UTR、CDS 和 3′ UTR 区域总体 m6A 修饰的百分比 (%)。
b 比较了 HeLa 细胞、水稻和拟南芥中分布在 5′ UTR、CDS 和 3′ UTR 区域的总体 m6A 修饰比例。分别整合了水稻和拟南芥所有组织的 m6A 位点,以计算各区域的总体 m6A 比例。
c 和 d 柱状图显示了水稻八种组织(c)和拟南芥九种组织(d)中组织特有和共同的 m6A 位点数量。m6A 位点数量是每个组织的两个生物学重复间的重叠位点。
e 元基因分布图展示了拟南芥九种组织中共同的 m6A 位点在转录本中的分布。
f 拟南芥九种组织中组织特有的 m6A 位点的元基因分布图。
g 元基因分布图展示了水稻中生殖组织特有、营养组织特有以及共同的 m6A 位点在转录本中的分布。花、种子和果荚被归类为生殖组织,其余组织归类为营养组织。每个转录本分为三个区域:5′ UTR、CDS 和 3′ UTR。图中标注了 m6A 位点数量。
组织特异性和共享 m6A 修饰
不同组织中的 m6A/A 比例和 m6A 位点数量各不相同,表明植物中存在组织共享和组织特异性的 m6A 修饰沉积。因此,我们分析了水稻和拟南芥所有组织中的组织特异性和共享的 m6A 修饰位点,分别鉴定出 14,953 个和 6,815 个所有组织共享的 m6A 位点(图 2c, d)。这些共享的 m6A 修饰位点主要富集在 3′ UTR 区域,而非 CDS 或 5′ UTR 区域,这在水稻(补充图 3d, e)和拟南芥(图 2e, f)中均有体现。这表明 3′ UTR 区域的 m6A 位点可能在整个生命周期中对植物转录组代谢具有普遍作用。
为了探索生殖组织和营养组织特有 m6A 位点的生物学差异,我们将不同组织划分为生殖组织和营养组织(参见方法)。分别在水稻和拟南芥中鉴定了生殖组织特有和营养组织特有的 m6A 位点(补充数据 1)。元基因分布图显示,与营养组织特有的 m6A 位点相比,生殖组织特有的 m6A 位点在 3′ UTR 区域的分布增加,表明 3′ UTR 区域的 m6A 调控在生殖阶段的重要性(图 2g 和补充图 3f)。GO 富集分析表明,水稻和拟南芥中包含生殖组织特有 m6A 位点的基因在类似的通路中富集,例如生殖结构发育、胚胎发育、免疫反应、光合作用和叶绿体组织(补充图 3g)。而包含营养组织特有 m6A 修饰的基因主要富集在刺激反应通路中,例如涉及水稻激素刺激反应的 ARF1、ARF7 和 ARF9 基因(补充图 3h),但这些通路在拟南芥中并不显著富集。这可能表明组织特异性的 m6A 甲基化在植物生长调控中发挥调节作用。
水稻和拟南芥中 m6A 调控的进化保守性和可变性
我们进一步研究了拟南芥和水稻直系同源基因对中 mRNA m6A 修饰的进化保守性。结果发现,在直系同源基因中共有 12,359 对保守的 m6A 位点(补充数据 2),而仅在拟南芥和水稻中分别鉴定出 108,856 个和 226,673 个特有 m6A 位点(图 3a)。有趣的是,在水稻(补充图 4a)和拟南芥(补充图 4b)中,特有 m6A 位点的平均 m6A 比例显著高于水稻-拟南芥保守位点。在保守 m6A 位点对中,7,734 对位于相同基序序列中(补充数据 3,补充图 4c 和图 3b)。保守 m6A 位点在同源基因中的比例相关性较弱(图 3c),且在不同组织中变化明显(图 3d 和补充图 4d),表明即使这些位点在所有组织中是保守的,其修饰水平仍具有组织依赖性。
鉴于 m6A 的存在对植物正常发育至关重要,我们进一步探索了水稻-拟南芥保守 m6A 位点的功能意义。GO 分析显示,包含保守 m6A 位点的基因显著富集于刺激响应和植物发育相关通路,例如叶绿体、光合作用、光形态建成、胚胎发育、茎发育、花发育、叶发育、根发育和胚珠发育(图 3e)。例如,与光合作用相关的基因 CAB3、Lhca5、LHCA3 和 LHB1B2;与根表皮细胞分化和根毛细胞分化相关的基因 POM1、GN、UBC36、GCS1、UBC35、GEM、SCN1 和 MRH1;以及与花发育相关的基因 PFT1、PS1、RDR6、DCL4、ARF8 和 MET1,它们在水稻和拟南芥中均具有保守的 m6A 修饰位点。
总体而言,这些结果为未来研究 m6A 在植物进化压力下的潜在作用提供了基础。
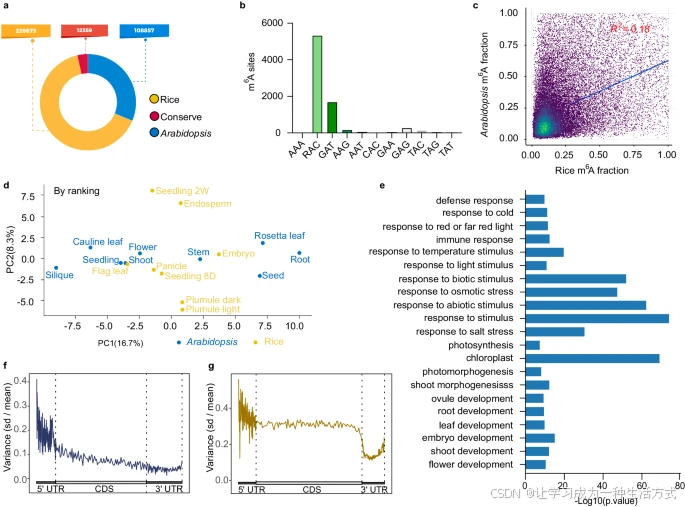
a 水稻和拟南芥之间保守和物种特有 m6A 位点的数量。
b 在保守 m6A 位点中,RAC 基序是最普遍的基序。基序序列分为三个组:RAC、GAT 和其他。其中碱基 "R" 表示 A 或 G。
c 水稻和拟南芥直系同源基因对中 m6A 比例的相关性分析。图中标注了 R² 值。
d 水稻和拟南芥所有组织中保守 m6A 位点的主成分分析(PCA)。这些 m6A 位点的修饰水平根据各自物种中的组织排名进行了标准化。
e 含有保守 m6A 位点的基因的基因本体(GO)分析。使用单侧 Fisher 精确检验,调整后的 P 值通过线性逐步提升方法计算。
f, g 显示水稻(f)和拟南芥(g)不同组织中 m6A 水平标准差与平均值之比(SD/Mean)的柱状图,分别针对 5′ UTR、CDS 和 3′ UTR 区域。总体而言,从 5′ UTR 到 3′ UTR,两种物种的方差均有所减少。特别是,水稻在整个基因结构中表现出逐步降低的方差,而拟南芥在 CDS 区域方差较为稳定,但在 3′ UTR 中方差急剧减少。
尽管水稻和拟南芥中存在进化保守的 m6A 修饰位点,但元基因分布图显示了某些差异,特别是在 3′ UTR 区域的 m6A 位点分布方面。为说明这些差异,我们分别整合了水稻和拟南芥不同组织中的所有 m6A 位点,并计算 m6A 位点数量的标准差与平均值之比(SD/Mean)。总体上,我们观察到在水稻(图 3f)和拟南芥(图 3g)中,从 5′ UTR 到 3′ UTR 的方差逐渐减少,而在 5′ UTR 区域的方差较为波动(图 3f, g)。尽管具有相似性,水稻表现出基因结构中逐步减少的方差,而拟南芥在 CDS 区域方差较为稳定,随后在 3′ UTR 中急剧减少(图 3f, g)。这些结果表明,在 CDS 和 3′ UTR 区域,水稻和拟南芥可能具有不同的 m6A 沉积调控机制。
植物基因组中 m6A 沉积的不同模式
早期研究已经阐明了哺乳动物和植物基因组中 m6A 的分布模式,表明 m6A 主要富集于最后一个外显子和长的内部外显子。这种分布受到外显子结构的影响,并由外显子连接复合体(EJC)调控。我们将这些分布模式归纳为三个基本规则:“长外显子规则”、“最后外显子规则”和“外显子结构规则”。利用单碱基分辨率的 m6A 数据,我们研究了这些规则在植物界中的保守性。
需要注意的是,人类基因组中的外显子通常比水稻和拟南芥中的外显子更长,这可能导致每个外显子和每个滑动窗口内 m6A 分布的差异。为提高测量精度,我们引入了两个指标:“m6A 密度”,即将每个外显子内的总 m6A 水平按外显子长度归一化;以及“m6A 可能性”,即将每个滑动窗口内的 m6A 水平按该窗口中外显子累积覆盖深度归一化。
我们重新分析了哺乳动物样本(尤其是 HeLa 细胞系)的单碱基 mRNA m6A 数据,并与水稻和拟南芥的数据进行比较。结果发现,当外显子长度小于 1000 个核苷酸(nt)时,无论在人类还是植物基因组中,m6A 水平通常随着内部外显子长度的增加而增加。然而,这种模式在人类基因组中长度超过 1000 nt 的内部外显子中不成立。水稻和拟南芥不显示这种趋势,主要因为它们几乎没有超过 1000 nt 的内部外显子。
在研究“m6A 密度”时,我们观察到无论在人类还是植物中,m6A 密度与内部外显子长度呈负相关。这表明 m6A 修饰的累积速度慢于外显子长度的增加。有趣的是,植物中的总体 m6A 密度高于人类,在外显子长度约为 100 nt 时达到峰值。这表明,在植物中,过长或过短的外显子可能更有效地降低 m6A 修饰的水平。
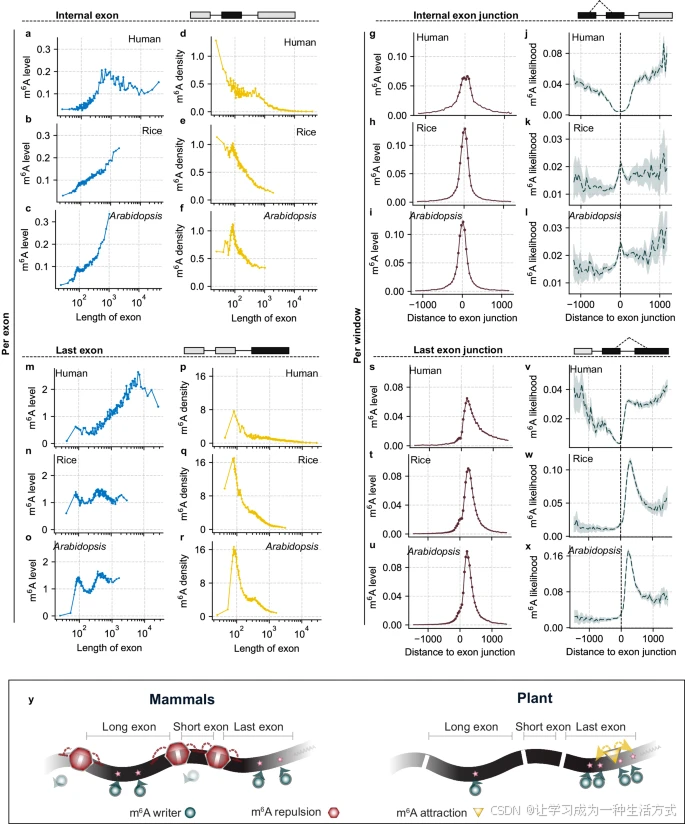
a-c 在人类(a)、水稻(b)和拟南芥(c)的转录本中,根据内部外显子的长度将其分为 100 等分组,并将每组的平均 m6A 水平(蓝点)与外显子长度绘制在一起。
d-f 显示了人类(d)、水稻(e)和拟南芥(f)转录本中每组内部外显子的“m6A 密度”与外显子长度的关系(黄色点)。m6A 密度通过将每个外显子中的 m6A 水平归一化为其长度后乘以 1000 计算得出。
g-i 所有内部外显子在其外显子连接位点对齐后,显示了在人类(g)、水稻(h)和拟南芥(i)基因组中滑动窗口内的整体 m6A 水平与外显子连接位点的距离关系(棕色点)。
j-l 在人类(j)、水稻(k)和拟南芥(l)转录本中显示了靠近内部外显子连接位点的“m6A 可能性”(深绿色线),阴影表示 95% 置信区间。
m-o 类似于面板 a-c,但显示了在人类(m)、水稻(n)和拟南芥(o)基因组中最后一个外显子的平均 m6A 水平与外显子长度的关系。
p-r 类似于面板 d-f,但显示了在人类(p)、水稻(q)和拟南芥(r)基因组中最后一个外显子的“m6A 密度”。
s-u 类似于面板 g-i,但显示了在人类(s)、水稻(t)和拟南芥(u)基因组中靠近最后一个外显子连接位点的 m6A 水平。
v-x 类似于面板 j-l,但显示了在人类(v)、水稻(w)和拟南芥(x)基因组中靠近最后一个外显子连接位点的“m6A 可能性”。
y 示意图展示了在人类中抑制模式与植物中激活模式对不同 m6A 分布模式的贡献。
在 j-l 和 v-x 中,数据以中位数值表示。
估算 m6A 修饰的概率
我们将所有外显子在其连接位点对齐,并计算了人类和植物滑动窗口内的 m6A 水平。靠近连接位点的区域通常外显子覆盖率较高,导致 m6A 水平增加(图 4g-i)。然而,当通过覆盖率归一化后(补充图 5q-s),在人类 HeLa 细胞系中,“m6A 可能性”在连接位点处表现出抑制行为,与先前的哺乳动物研究一致。相比之下,这种模式在水稻和拟南芥中并未观察到,反而显示出轻微的相反趋势。这种差异表明,EJC 复合体可能在哺乳动物细胞中被动抑制 m6A 沉积,而在植物中则未发生类似情况。相反的趋势提出了一个有趣的问题,即“外显子结构”规则是否在植物中普遍保守。活跃的 m6A 沉积途径可能塑造了某些植物中 mRNA 的 m6A 分布。
关于最后一个外显子
在人类 HeLa 细胞系中,即使外显子长度超过 1000 nt,m6A 水平与外显子长度之间的正相关关系依然存在(图 4m 和补充图 5k)。这种相关性在水稻和拟南芥中明显不存在(图 4n, o 和补充图 5l, m)。此外,在人类和植物中,最后一个外显子的“m6A 密度”与外显子长度呈负相关(图 4p-r 和补充图 5n-p)。与内部外显子相比,人类和植物中最后一个外显子的 m6A 更集中于长度约为 100 nt 的外显子中。值得注意的是,水稻和拟南芥最后一个外显子的“m6A 密度”高于人类,即使总体 m6A 水平较低。这些发现表明,尽管最后一个外显子中 m6A 富集的总体模式在进化上是保守的,但这种修饰与外显子长度之间的关系在不同物种中存在显著差异。
在靠近最后一个外显子连接位点的“m6A 可能性”方面,我们在人类和植物中都观察到连接位点下游的不对称峰值(图 4s-u 和补充图 5t-v)。与内部外显子类似,在哺乳动物中,“m6A 可能性”在靠近最后外显子连接点时逐渐降低(图 4v)。值得注意的是,这种分布模式在连接位点处出现了特定的突变点(图 4j, v)。这表明,在哺乳动物中,EJC 复合体在抑制 m6A 沉积中起到了显著作用,m6A 的总体分布受到被动过程的调控。然而,水稻和拟南芥在最后外显子连接位点下游约 300 nt 处表现出明显的峰值,然后逐渐减少(图 4w, x)。这可能表明植物中存在活跃的 m6A 沉积过程,增强了 m6A 甲基转移酶在该区域的招募。
由于大多数终止密码子位于最后一个外显子中,我们基于它们与终止密码子的距离对外显子进行对齐以进行更细致的分析。我们观察到水稻和拟南芥在靠近终止密码子附近有显著的突变点,并发现 m6A 可能性的峰值比最后外显子起点更接近终止密码子(补充图 5w-y)。这些发现表明,植物中 m6A 的活跃沉积可能受到终止密码子附近特定基因组特征的影响(图 4y)。综合来看,这些研究表明植物和哺乳动物之间的 m6A 修饰机制存在潜在的显著差异,提示植物 mRNA m6A 沉积可能存在尚未探索的新机制。
m6A 修饰主要通过 3′ UTR 增强拟南芥幼苗中的 mRNA 稳定性和翻译
m6A 修饰主要通过 3′ UTR 位点促进哺乳动物 mRNA 的周转。与哺乳动物相比,m6A 在植物中的作用尚不清晰,而一些研究表明 m6A 修饰能够稳定拟南芥中的修饰 mRNA。为获得 m6A 水平与 mRNA 周转之间更准确的关系,我们检索了拟南芥幼苗中公开的 RNA 半衰期数据,发现 m6A 修饰的转录本往往比未修饰的转录本具有更长的寿命。植物中存在偏好结合 m6A 修饰 mRNA 的冗余 ECT 蛋白。我们进一步结合公开的 ECT2 CLIP-seq 数据和 mRNA 半衰期数据,研究了 ECT2 调控的 mRNA 降解效应。我们观察到 m6A 位点与 ECT2 靶点之间有大量重叠,确认了 m6A 被 ECT2 结合。包含 m6A 位点的 ECT2 靶基因的寿命高于未被 ECT2 结合的基因,表明 m6A 能够稳定 mRNA,而 m6A 识别蛋白可以增强拟南芥幼苗中 mRNA 的稳定性。
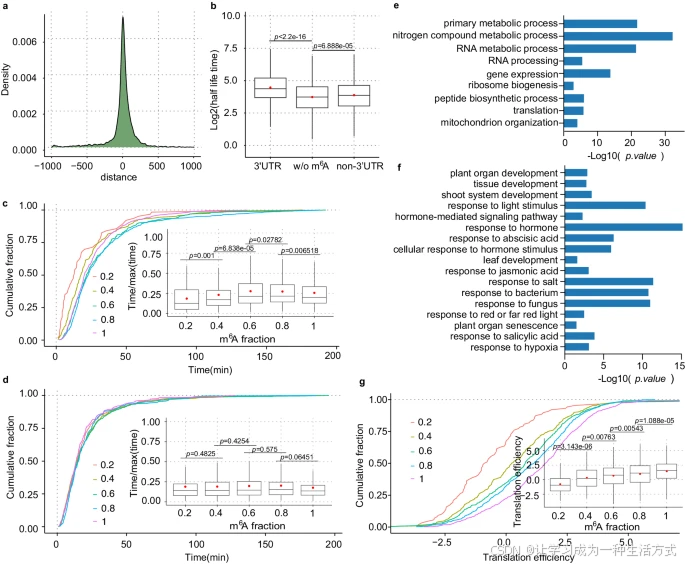
a. 密度图显示通过CLIP-seq鉴定的ECT2靶点的峰中心与通过m6A-SAC-seq在拟南芥幼苗中鉴定的m6A位点之间的距离分布。
b. 比较仅在3′ UTR区域发生m6A修饰、非3′ UTR区域发生m6A修饰以及没有m6A修饰的转录本的寿命差异。3′ UTR,n=1,813;非3′ UTR,n=2,645;无m6A修饰,n=3,319。
c. 累积曲线和箱线图显示仅在3′ UTR区域发生m6A修饰的转录本的mRNA寿命分布。根据m6A比例总和将转录本分为五类:(0,0.2),n=62;(0.2,0.4),n=228;(0.4,0.6),n=353;(0.6,0.8),n=483;(0.8,1),n=612。
d. 累积曲线和箱线图显示在非3′ UTR区域发生m6A修饰的转录本的mRNA寿命分布。根据m6A比例总和将转录本分为五类:(0,0.2),n=466;(0.2,0.4),n=981;(0.4,0.6),n=566;(0.6,0.8),n=280;(0.8,1),n=294。
e. 针对与仅在3′ UTR区域发生m6A位点相关的基因进行GO富集分析。
f. 针对与非3′ UTR m6A位点相关的mRNA进行GO富集分析。e和f中使用单侧Fisher精确检验,调整的p值通过线性逐步上升法计算。
g. 仅在3′ UTR区域发生m6A修饰的转录本表现出与翻译效率的强正相关。根据m6A比例总和将转录本分为五类:(0,0.2),n=88;(0.2,0.4),n=293;(0.4,0.6),n=489;(0.6,0.8),n=620;(0.8,1),n=749。c、d和g中,使用了拟南芥幼苗的mRNA衰变数据集GSE86361进行mRNA寿命分析,并使用了数据集GSE206292进行翻译效率分析。b-d和g中p值通过单尾Wilcoxon秩和检验确定。箱线图中,中心线表示中位数,红点表示均值,上下四分位数为箱体边界。源数据提供于源数据文件中。
我们随后研究了m6A修饰位置是否可能影响mRNA稳定性差异,如哺乳动物中观察到的那样。为此,我们首先将m6A位点聚类为仅在3′ UTR区域的m6A和非3′ UTR区域的m6A,发现仅在3′ UTR区域的m6A显著稳定了mRNA,而非3′ UTR区域的m6A仅导致与无m6A修饰基因相比的轻微寿命增加(图5b)。这表明仅在3′ UTR区域的m6A在调控mRNA稳定性,尤其是稳定修饰的转录本中具有更主要的作用。
为进一步探讨m6A比例水平与mRNA稳定性的关系,我们将携带仅在3′ UTR区域和非3′ UTR区域m6A位点的mRNA分为五组,基于m6A比例发现,较高的m6A比例与携带仅在3′ UTR区域m6A位点基因的较高mRNA稳定性相关(图5c),而携带非3′ UTR区域m6A位点的基因中未观察到显著相关性(图5d和补充图6c)。上述结果通过Sorenson, R. S等人的mRNA代谢数据进一步证实(补充图6d、e)。尽管不同的读取蛋白可识别不同区域的m6A以发挥稳定或衰变功能,但由于大多数mRNA的m6A修饰集中在3′ UTR区域(图2a、b),这可能表明m6A在植物中整体具有mRNA稳定化效应。
m6A修饰位置对控制mRNA稳定性的不同影响促使我们探讨m6A位置是否决定了生物学功能的差异。GO富集分析表明,与仅在3′ UTR区域的m6A相关的基因显著富集于基因表达、RNA加工和核糖体生物合成等一般生物学通路(图5e),而与非3′ UTR m6A相关的mRNAs富集于激素反应、真菌反应、盐胁迫反应、叶片发育和植物器官衰老等更具体的通路(图5f)。此外,我们还观察到仅在3′ UTR区域发生m6A修饰的转录本中,m6A水平与翻译效率呈正相关(图5g),而在携带非3′ UTR区域m6A位点的转录本中未观察到显著相关性(补充图6f)。这些观察表明,植物中m6A的多样化作用受其位置及其下游结合蛋白的影响。
MTA在拟南芥幼苗叶绿体转录组中的m6A修饰
接下来,我们在拟南芥mta突变体幼苗(补充图6g、h)中利用m6A-SAC-seq分析了m6A位点,并将其与野生型(WT,Col)幼苗的m6A位点进行了比较。在WT中共鉴定出2,894个RNA上的14,125个MTA依赖性m6A位点。这些m6A位点在mta中的甲基化水平显著降低(10,505个位点)或完全丧失(3,621个位点);这些m6A位点被定义为MTA依赖性m6A位点。我们注意到,MTA依赖性m6A位点更倾向于位于3′ UTR区域而非CDS和5′ UTR区域(图6a)。GO富集分析显示,含有这些m6A位点的mRNAs大多富集于刺激响应、叶绿体、光合作用膜和胚后发育(图6b)。这与之前关于MTA参与盐胁迫反应、蓝光响应和胚胎发育的研究结果一致。值得注意的是,叶绿体相关转录本(538个基因)和光合作用膜相关转录本(59个基因)的m6A水平在mta中显著降低,其中20个m6A位点在叶绿体转录组中显示出m6A水平的显著下降(图6c)。叶绿体编码转录本的甲基化水平在不同组织中有所变化(图6c),表明拟南芥生命周期中叶绿体转录组的MTA依赖性m6A修饰是动态的。
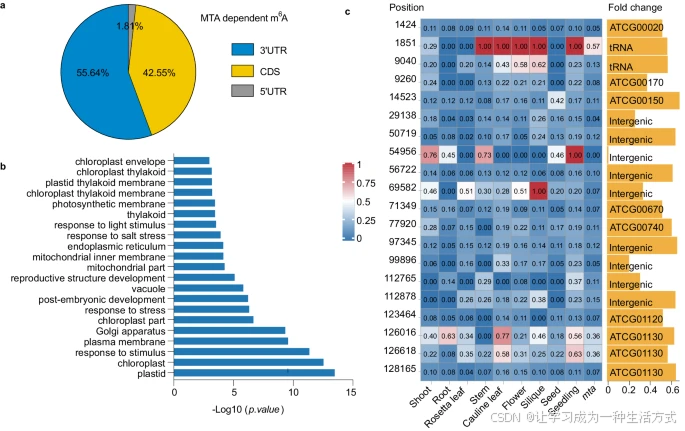
a. MTA依赖性m6A位点在拟南芥幼苗转录本上的分布。 b. 包含MTA依赖性m6A位点的mRNA的GO富集分析。单侧Fisher精确检验,调整的P值使用线性逐步上升法计算。 c. 叶绿体编码基因在不同组织中的甲基化水平热图,显示叶绿体编码基因上m6A位点的位置,同时展示m6A水平及其变化倍数。源数据可在源数据文件中获取。
接下来,我们利用已发表的数据集(补充图6i)观察到,mta突变体的整体翻译效率相较于野生型(WT)显著降低。然而,相较于WT对照,mta突变体中翻译效率上调(332个基因,倍数变化 >2,p < 0.05)和下调(257个基因,倍数变化 <0.5,p < 0.05)的基因数量相似。MTA通过安装m6A对翻译效率的影响在拟南芥幼苗中表现为异质性,与哺乳动物中观察到的情况相似。或许一致的是,这些翻译效率差异的基因也显著富集于不同的GO分类中。翻译效率上调的基因主要富集于核糖体和核仁等一般途径(补充图6j),而翻译效率下调的基因则特异性富集于叶绿体和光合作用膜(补充图6k),如FIBRILLIN 4 (FIB4)和SMO2基因。总体而言,MTA在核和叶绿体转录组中均沉积m6A修饰,从而调控光合作用。
光诱导的m6A对生物钟的反馈调控
m6A修饰对mRNA的甲基化在植物和哺乳动物中均调控生物钟。为进一步探究光对水稻m6A甲基化的影响,我们分别在黑暗(每天24小时黑暗)和光照条件下(每天16小时光照,8小时黑暗)萌发水稻种子。使用m6A-SAC-seq分析了在黑暗和光照条件下萌发3天后的胚芽(图7a)。我们的数据揭示,与黑暗条件相比,光照条件下m6A甲基化水平普遍升高(23,253个m6A位点超甲基化,2,607个位点低甲基化,补充图7a-c)。光诱导的m6A位点主要位于3′ UTR之外(图7b)。GO富集分析表明,包含超甲基化m6A位点的基因显著富集于刺激响应途径,包括光刺激和激素刺激(补充图7d)。值得注意的是,光显著增加了光受体转录本的m6A修饰水平,例如PHYA (Chr3:29172686; CDS; AGATA),PHYB (Chr3:11021272; CDS; AGATA),PHYC (Chr3:31007707; CDS; GGACA),CRY1a (Chr2:21976854; 5′ UTR; AGAGC),和CRY2 (Chr2:24921916; 3′ UTR; AAACT)。因此,与哺乳动物中观察到的情况类似,水稻生物钟基因在其转录本内的m6A甲基化水平因光而发生显著变化。
此外,在上述拟南芥mta突变体中,CRY1 3′ UTR区(Chr4:5727183; AAACA; 3′ UTR)的m6A比例降低,可能导致翻译效率下降,这表明MTA通过m6A沉积调控CRY1的翻译(图7c, d)。鉴于光诱导的CRYs相分离可以调控植物中MTA的活性,且MTA通过对CRY转录本的m6A修饰控制其翻译,似乎在植物中存在一个环状的表观转录组-翻译对生物钟的反馈调控机制。
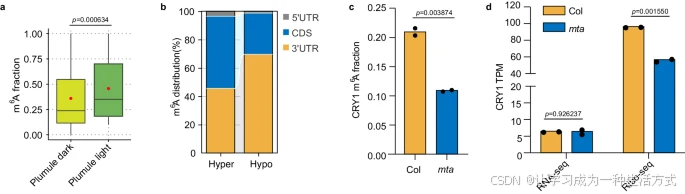
a. 光照增加了水稻胚芽中的m6A比例。数据以均值 ± 标准差表示,n=2。p值通过单尾Wilcoxon秩和检验计算。 b. m6A位点在5′ UTR、CDS和3′ UTR区域的相对分布。 c. 在mta突变体中观察到CRY1的m6A比例降低。数据为均值,n=2。 d. mta突变体中CRY1的翻译效率降低。翻译效率数据集GSE206292用于分析。数据为均值,n=2。统计学差异通过Student t检验确定。源数据提供于源数据文件中。
讨论
mRNA的m6A甲基化在植物和哺乳动物的发育以及信号传递和刺激响应中起着关键作用。以往植物中关于RNA m6A的研究缺乏碱基分辨率、精确性以及修饰计量比的信息。得益于首次在哺乳动物转录组中应用m6A-SAC-seq方法实现的单碱基分辨率m6A测序技术,我们在此报告了覆盖水稻和拟南芥生命周期的8种水稻组织和9种拟南芥组织的具有计量比信息的m6A单碱基精确图谱。这些图谱提供了高置信度的单碱基分辨率m6A位点,为未来研究水稻和拟南芥中m6A的功能提供了深入资源。
我们鉴定了拟南芥和水稻直系同源基因对中进化保守的m6A修饰位点。包含保守m6A位点的直系同源基因对在组织发育、光合作用和刺激响应中具有重要作用,并可能受到进化压力的选择。尽管这些直系同源基因中存在保守的m6A位点,我们仍观察到水稻和拟南芥在3′ UTR中m6A分布模式的差异。此外,尽管总m6A水平与内含子长度呈正相关,但不同于哺乳动物,在植物中未观察到与最后一个外显子长度的相关性。哺乳动物的mRNA m6A分布似乎是通过外显子连接复合体或其他蛋白因子介导的抑制通路塑造的,而植物则依赖于主动安装过程控制m6A沉积。特别是,我们的结果表明植物mRNA中接近终止密码子的区域存在主动m6A沉积过程。这种抑制-激活双重沉积调控可能解释了不同物种中m6A分布模式的差异。我们和其他研究已经表明,m6A抑制在哺乳动物中由EJC介导,而这种抑制-激活模型预测了RNA结合蛋白可能通过招募m6A写入蛋白在植物mRNA的终止密码子附近引导m6A沉积。这些观察表明,植物转录组中可能存在一种未知的m6A分布调控机制,需要进一步深入研究。
从我们的分析来看,大多数植物m6A修饰集中在3′ UTR区域。植物中的研究表明m6A修饰能够稳定mRNA,而在哺乳动物中,m6A修饰倾向于使修饰的mRNA不稳定。基于我们对拟南芥幼苗的单碱基分辨率结果,我们观察到3′ UTR区域m6A位点的甲基化水平与mRNA稳定性呈正相关,验证了m6A在某些植物组织中对mRNA的整体稳定作用。一致地,ECT2蛋白结合的m6A位点表现出显著延长的半衰期,相比之下,未被ECT2结合的转录本半衰期较短,这表明植物中m6A结合蛋白可以增强mRNA稳定性。此外,MTA安装的m6A修饰可以在特定途径中促进或降低翻译效率,这与哺乳动物中的观察结果类似。因此,m6A修饰通过结合蛋白稳定修饰的转录本,同时在拟南芥幼苗中表明一种整体翻译促进效应。
总之,这些跨越水稻和拟南芥生命周期的基于碱基分辨率和定量的m6A修饰图谱填补了植物研究中的一项显著空白。人类和植物之间单碱基m6A图谱的比较分析揭示了在不同物种中塑造m6A分布模式的抑制-激活双重调控模型。
方法
植物材料
本研究使用了拟南芥(Arabidopsis thaliana)Col-0品系和水稻(Oryza sativa)粳稻品种日本晴(Nipponbare)。
-
拟南芥植株在22 °C、每日16小时光照条件下生长。
-
拟南芥幼苗在1/2 Murashige和Skoog(MS)培养基上培养7天后采集。
-
拟南芥的地上部分和根在MS培养基上培养14天后采集。
-
拟南芥的莲座叶在土壤中培养30天后采集。
-
拟南芥的苞叶、花、茎和角果在开花后采集。
-
拟南芥的种子在完全干燥后采集。
-
拟南芥mta突变体(ABI3::MTA/mta)的种子在MS培养基上播种,并在22 °C、每日16小时光照条件下培养8天后采集幼苗。
-
-
水稻植株在28 °C、每日14小时光照条件下生长。
-
水稻的8天龄和2周龄幼苗被采集。
-
水稻抽穗期的穗状花序、开花后10天的旗叶、胚乳和胚被采集。
-
光照和黑暗条件下,萌发3天的胚芽被采样。
-
所有组织均快速冷冻于液氮中,使用研钵和杵研磨,并储存于−80 °C。总RNA通过TRIzol™试剂(目录号:15596026)按照制造商说明提取。所有植物均在香港中文大学的温室中种植。
从总RNA中捕获mRNA
每个生物学重复使用50 μg总RNA用于mRNA捕获,使用Dynabeads mRNA DIRECT Purification Kit(Invitrogen)按照制造商说明并略作修改。
-
将50 μg总RNA用100 μl水稀释后,于65 °C变性2分钟,并立即放入冰上冷却2分钟。
-
用200 μl裂解/结合缓冲液将100 μl的Dynabeads洗涤两次,然后在100 μl裂解/结合缓冲液中洗脱。洗脱的磁珠与变性的总RNA混合后,室温下在旋转装置上结合15分钟。
-
用洗涤缓冲液B洗涤两次后,用30 μl水洗脱磁珠,并立即于75 °C孵育2分钟。
-
经磁分离后收集洗脱的mRNA,重复以上过程以获得更纯化的mRNA。
通过LC–MS/MS量化RNA中的m6A
50 ng mRNA被消化为核苷,并通过Agilent 6460 Triple Quad MS–MS与1290 UHPLC系统(配备ZORBAX Eclipse XDB-C18柱)测量m6A的含量,并基于纯标准品生成的标准曲线计算含量。
-
样品用nuclease P1(NEB)在37 °C下消化2小时。
-
加入1 μl虾碱性磷酸酶(rSAP)和3 μl 10× rCutsmart缓冲液(NEB),在37 °C孵育2小时。
-
样品通过0.22 μm过滤器(Millipore)过滤并注入LC–MS/MS。
-
核苷通过核苷-碱基离子质量转移(m6A:282到150,A:268到136)定量。
-
使用同一批样品运行的纯核苷标准品生成的标准曲线进行量化,计算m6A与A的比例。
m6A-SAC-seq文库构建
每个重复使用50 ng mRNA构建文库。所有文库完全按照之前发表的协议进行构建。构建的文库在Illumina HiSeq测序平台上以双端模式(每读长150 bp)进行测序。
m6A-SAC-seq数据处理
测序完成后,通过(GitHub: m6A-SACseq)检测m6A位点。分析使用从Ensembl数据库下载的参考基因组,其中拟南芥使用TAIR10版本,水稻使用IRGSP-1.0版本。
RNA寿命分析和翻译数据分析
拟南芥Col幼苗的RNA寿命和翻译效率数据下载自数据集GSE206292和GSE118462。拟南芥mta突变体的翻译效率数据同样来自GSE206292。
水稻与拟南芥直系同源基因中的保守m6A位点
首先鉴定水稻和拟南芥之间的一对一直系同源基因。通过成对比对获得具有一致侧翼序列(±1 nt)并以A位点为中心的同源位置。上述A位点定义为保守A位点,而保守A位点上的m6A修饰定义为保守m6A位点。
基因本体(GO)分析
通过农业社区网络工具agriGO v2.0(agriGO)进行功能GO富集分析。假发现率(FDR) < 0.05的GO术语被认为显著富集。
统计和重复性
所有实验独立重复至少两次,并显示相似结果。图形绘制使用GraphPad Prism v.9和R studio完成。


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









