










The pineapple reference genome: Telomere-to-telomere assembly, manually curated annotation, and comparative analysis
菠萝参考基因组:端粒到端粒组装、人工精修注释和比较分析

摘要
菠萝是全球第三大重要的热带水果,共有五个品种。目前,不同菠萝品种的基因组已被发布,然而,它们均不完整,存在大量基因组空缺,并且仅涵盖五个品种中的两个。这极大地阻碍了菠萝育种工作的进展。在本研究中,我们对三个菠萝品种的基因组进行了测序,包括一个野生菠萝品种、一个纤维菠萝品种和一个全球广泛种植的食用菠萝品种。我们构建了首个无缺口的菠萝参考基因组(Ref)。通过整合多种证据来源,并对每个基因结构注释进行人工修订,我们鉴定出了 26,656 个蛋白编码基因。BUSCO 评估结果显示,该基因组的完整性达 99.2%,证明了其高质量的基因结构注释。利用这些数据资源,我们在三个品种中鉴定出 7,209 个结构变异(SVs)。约 30.8% 的菠萝基因位于结构变异 ±5 kb 范围内,其中包括 30 个与花青素合成相关的基因。进一步的分析和功能实验表明,AcMYB528 的高表达与叶片中花青素的积累相一致,这一现象可能受到 1.9-kb 插入片段的影响。此外,我们开发了 Ananas 基因组数据库,提供数据浏览、检索、分析和下载功能,填补了菠萝基因组资源数据库的空白。综上,我们获得了一个无缝衔接的高质量菠萝参考基因组,为菠萝基因组学研究奠定了坚实基础,并为菠萝育种提供了重要的参考。
引言
菠萝 (Ananas comosus (L.) Merr.) 属于凤梨科(Bromeliaceae),在进化生物学中占据重要地位,因此成为植物学研究的一个重要课题 (Coppens d'Eeckenbrugge and Leal, 2003; Coppens d'Eeckenbrugge and Govaerts, 2015)。不同品种的菠萝具有不同用途:部分品种以其鲜果风味受到青睐,部分品种因其高纤维含量而被利用,另一些品种则具有观赏价值。每个品种均具有独特的性状,将这些性状结合以满足工业生产需求和消费者偏好的多样化,是当前菠萝育种和改良工作的核心目标。
近年来,多个菠萝品种的基因组已被测序,为菠萝基因组学研究带来了显著进展。最早被测序的品种是 F153 (Ming et al., 2015),随后是观赏菠萝 A. comosus var. bracteatus CB5 (Chen et al., 2019) 和栽培品种 Yugafu (Nashima et al., 2022)。尽管取得了这些进展,但已发布的基因组仍存在大量缺口,且基因结构注释不完整,阻碍了菠萝基因组学、遗传学和育种领域的进一步研究。
我们致力于培育既具观赏价值又能提供优质果实的栽培菠萝。在之前的研究中,我们鉴定了一些具有 红色叶片或果皮 的独特菠萝种质资源,这些性状源于 花青素的积累 (Luan et al., 2023)。通常,MYB 转录因子与 bHLH 和 WD40 蛋白相互作用,调控 CHS(查尔酮合酶)和 ANS(花色苷合成酶)等花青素生物合成通路中的基因表达。我们发现,在 红皮菠萝 基因组中存在 120-bp 的插入序列,这一插入显著提高了 MYB 基因在萼片中的表达水平,导致果皮呈红色 (Zhang et al., 2024)。基于这些发现,我们推测,对 结构变异(SVs) 进行全面分析,将有助于培育具有丰富色彩的栽培菠萝,例如 红叶品种。
由于尚无完整的菠萝参考基因组,我们对 三个代表性品种 进行了测序,以增强比较基因组学分析。这三个品种分别为:
-
“Ba Li” (BL):栽培菠萝(Queen)主要品种之一;
-
“Yu Ling Long” (YLL, A. comosus var. microstachys):观赏菠萝品种;
-
“Li Ye” (LY, A. comosus var. erectifolius):早期驯化菠萝品种。
其中,BL 是目前全球广泛种植的菠萝品种之一,与 F153 和 MD2 齐名 (Sun, 2011),以易种植、芳香浓郁和优良的储藏特性闻名。YLL 具有 极短的营养生长期(6 个月),远短于 F153 和 MD2(12 个月),因此在商业育种中具有重要价值。LY 具有 红色果皮和无刺红叶,在栽培周期、叶片特性和果实性状方面,与 F153、CB5 和 Yugafu 等品种存在显著差异。
在本研究中,我们利用 Oxford Nanopore Technologies(ONT)长读长测序、高保真(HiFi)测序 和 高通量染色体构象捕获(Hi-C)技术,组装了 首个完整无缺口的菠萝参考基因组。该基因组经过严格的人工精修,以确保高质量的基因结构注释。基于优化的基因组序列和注释信息,我们全面分析了菠萝中的 结构变异,并对调控 花青素积累 的 MYB 转录因子 进行了功能鉴定。此外,我们构建了 全面的菠萝基因组数据库,为菠萝基因组学和遗传学研究提供了宝贵资源。
结果
选定用于基因组测序的三种菠萝品种
菠萝共有 五个品种(图 1A),其中包括 两个野生品种(A. comosus var. microstachys 和 A. comosus var. parguazensis),以及 三个驯化品种(食用菠萝 A. comosus var. comosus、纤维菠萝 A. comosus var. erectifolius 和观赏菠萝 A. comosus var. bracteatus)(Coppens d'Eeckenbrugge and Leal, 2003; Coppens d'Eeckenbrugge and Govaerts, 2015)。截至目前,野生品种和纤维菠萝的基因组尚未被测序。
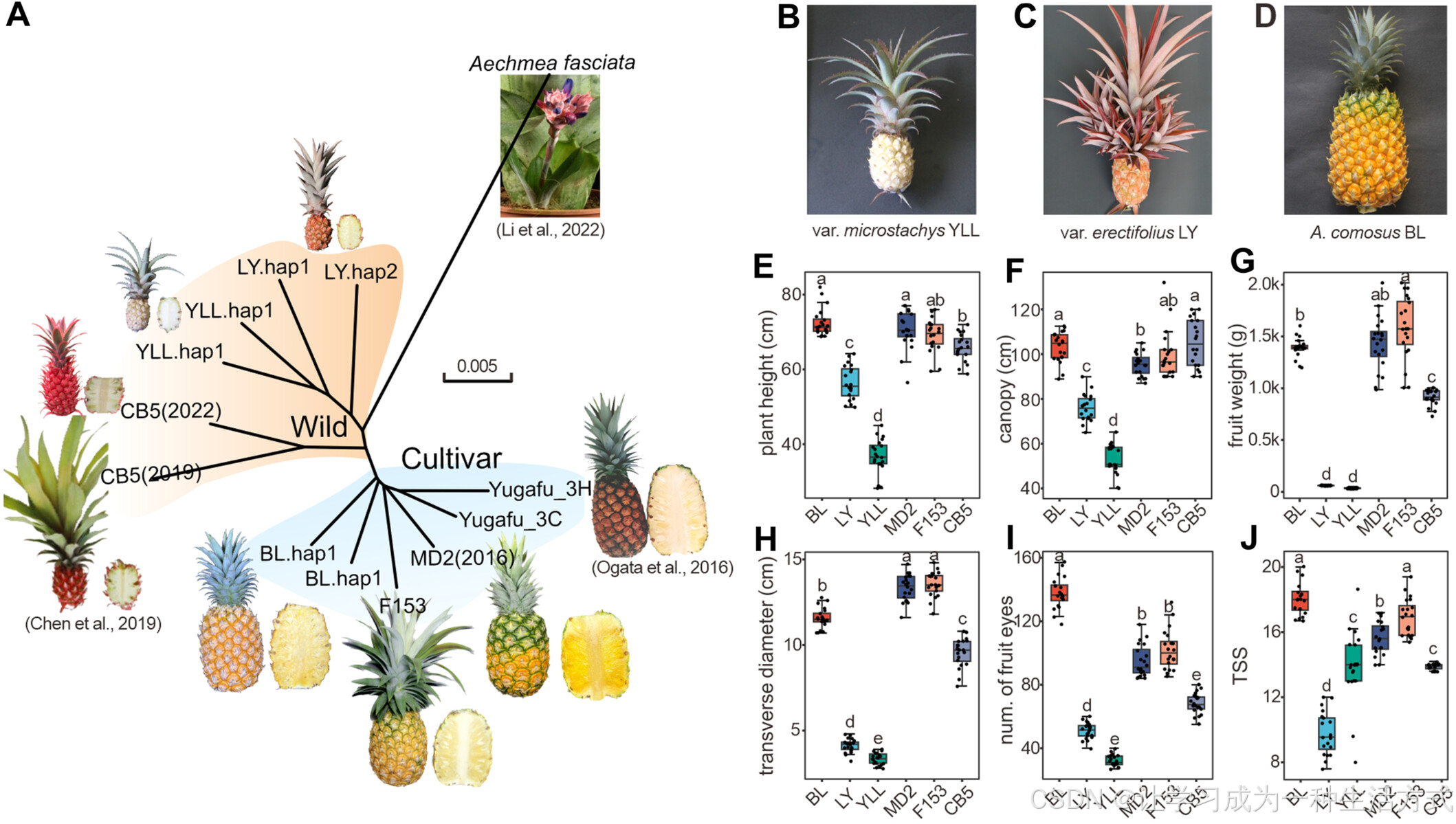
凤梨属物种的系统发育关系及其特征
(A) 系统发育树展示了本研究数据与已发布菠萝基因组数据的遗传关系。所有栽培菠萝属于同一分支,而其他品种则归入另一分支,以 Aechmea fasciata 作为外类群 (Li et al., 2022)。值得注意的基因组数据包括 CB5(2019)(Chen et al., 2019)、CB5(2022)(Feng et al., 2022)、以及 MD2 和 F153(Redwan et al., 2016;Ming et al., 2015)。此外,3H 和 3C 是 Yugafu 菠萝的两个单倍型(Nashima et al., 2022);Yugafu 的图像来源于 Ogata et al. (2016)。
(B–D) 代表性品种 “Yu Ling Long”(YLL)、“Li Ye”(LY) 和 “Ba Li”(BL) 的成熟果实及其顶冠形态。
(E–J) YLL、LY 和 BL 在植株高度、冠幅、果重、果横径、果眼数以及总可溶性固形物含量方面,与已发布基因组的品种(MD2(2016)、F153、CB5(2022))存在显著差异。箱线图显示了中位数,箱体边缘代表 25th 和 75th 百分位数,须线代表 最大和最小值(1.5 倍四分位距内)。显著性差异采用 Duncan 多重范围检验 进行分析。
A. comosus var. microstachys 作为一种 野生菠萝品种,因其 丰富的遗传多样性 被认为是栽培菠萝的共同祖先(Duval et al., 2001, 2003)。YLL(A. comosus var. microstachys)属于这一野生类型,叶片修长,具有 连续的刺状边缘,果实 小且呈乳白色(图 1B)。YLL 的 营养生长期极短(表 S1),使其成为 缩短商业品种生长周期的重要遗传资源。
纤维菠萝 “Li Ye”(LY,A. comosus var. erectifolius) 是一种 早期驯化品种,植株 生长直立,具有大量 分蘖和侧芽(表 S1)。与大多数 绿色栽培菠萝 不同,LY 具有 红色叶片、红色侧芽和红色果皮(图 1C),且 无刺,类似于栽培品种 F153。LY 与野生型 A. comosus var. microstachys 共享多个性状。历史上,LY 由于 含有约 6% 的干纤维含量(Leal and Amaya, 1991),曾被用于 吊床和渔网的生产。该品种体现了 土著居民的驯化过程,目前主要作为 观赏品种 进行栽培。
在 食用菠萝品种 中,Queen 群 是 哥伦布时代前 就已广泛分布的菠萝类型(Duval et al., 2001)。该群体与 Cayenne(卡宴)、Spanish(西班牙) 及 杂交组 共同构成了栽培菠萝的主要类别。BL 作为全球领先的品种,代表了 Queen 群。与基因组测序品种 Cayenne(F153) 和 杂交种(MD2) 相比,BL 具有 更小的顶芽、叶片边缘全缘有刺(图 1D),且货架期更长(Coppens d'Eeckenbrugge et al., 1997)。
综上所述,栽培品种 BL 和 LY 以及野生品种 YLL,与 MD2(2016)、F153 和 CB5(2022)在植株大小、果实重量和鲜果品质等方面存在显著差异(图 1E–J;表 S1)。然而,它们的 基因组仍未被深入研究。因此,我们选择这三个品种进行 基因组测序,以提供更 全面且高质量的遗传参考,为 菠萝育种 提供重要数据支持。
三种菠萝的端粒到端粒(T2T)基因组组装
我们获得了 BL、YLL 和 LY 的 HiFi 测序数据(平均覆盖度 约 65X)和 Hi-C 数据(平均覆盖度 约 118X)(表 S2)。结果显示 BL、YLL 和 LY 的基因组大小 分别为 369 Mb、359 Mb 和 365 Mb,且 杂合度分别为 1.45%、1.45% 和 2.23%,表明它们具有 高度杂合的基因组(图 S1)。我们采用 单倍型识别组装策略,结合 HiFi 和 Hi-C 数据 组装了 六个单倍型。每个单倍型的 Contig 数量在 184 至 695 之间,平均 375 个 Contig,Contig N50 值 介于 11.6 Mb 至 15.2 Mb(表 S3)。
为了验证组装的完整性,我们手动检查了 Contig 末端 是否含有植物基因组的 端粒重复序列模式(CCCTAAA)(图 S2)。在 所有六个单倍型中,共检测到 58 个 Contig 含有 双端端粒重复序列,平均 每个单倍型 9.7 个完整端粒到端粒(T2T)序列。
以 F153 基因组 作为参考,我们手动选择 最长的同源染色体 以构建 参考基因组(图 S2;表 S4)。最终得到 25 条染色体,总长度 423.8 Mb(表 S5)。参考基因组中的 重复序列 长度为 261 Mb,占基因组总大小的 61.6%(表 S6)。此外,长末端重复序列(LTR)组装指数(LAI) 值 达到 17.5(表 S7)。
尽管基因组中 重复序列比例较高,但参考基因组的 BUSCO 评分 以及 基因组注释完整性评分 分别达到了 99.3% 和 99.2%,表明其 基因组完整性极高(表 1、S8、S9)。
Table 1. Data statistics of genome assembly
| Yugufa | ||||||||
|---|---|---|---|---|---|---|---|---|
| Ref | MD2 (2016) | F153 | CB5 (2019) | CB5 (2022) | 3H | 3C | ||
| scaffold/chromosomes | No. of chromosomes | 25 | 8,448 | 25 | 25 | 25 | 25 | 25 |
| Total length of anchored (Mb) | 423.8 | 524.1 | 315.8 | 486.6 | 483.2 | 401.6 | 431.1 | |
| Anchored rate (%) | \ | \ | 82.7 | 94.8 | \ | \ | ||
| No. of gaps | 0 | 8,645 | 4,553 | 1,867 | 479 | 1,763 | 1,804 | |
| GC content (%) | 0.39 | 0.39 | 0.38 | 0.39 | 0.4 | 0.39 | 0.39 | |
| Maximum chr. length (Mb) | 20.99 | 1.29 | 17.33 | 27.13 | 31.89 | 23.88 | 33.04 | |
| Mean chr. length (Mb) | 16.95 | 0.06 | 12.63 | 19.46 | 19.33 | 16.07 | 17.25 | |
| Median chr. length (Mb) | 17.60 | 0.02 | 12.61 | 20.18 | 19.39 | 15.43 | 16.67 | |
| N50 (Mb) | 18.21 | 0.02 | 13.10 | 20.31 | 20.38 | 15.50 | 17.17 | |
| N base (bp) | 0 | 14,321,016 | 4,583,126 | 186,700 | 52,408 | 176,341 | 180,445 | |
| BUSCO | Genome BUSCO (%) | 99.3 | 98.3 | 98.2 | 95.1 | 98.6 | 91.4 | 96.0 |
| Proteome BUSCO (%) | 99.2 | 82.5 | 88.0 | 82.4 | 84.0 | 75.0 | 78.7 |
我们采用多种方法评估了参考基因组的组装质量。HiFi、ONT 以及来自不同菠萝品种的转录组的比对率分别为 99.98%、99.95% 和 88.35%(表 S10)。基于 k-mer 频谱评估基因组组装质量的 Merqury 工具(Rhie et al., 2020)对参考基因组的质量值(QV)评分为 66.52,表明 碱基错误率低于 10⁻⁶(表 S11)。此外,基于区域和结构错误评估基因组错误率的 Clipping reveals assembly quality(CRAQ) 工具(Li et al., 2023)计算得出 区域组装质量指标(R-AQI)和整体组装质量指标(S-AQI) 分别为 98.66 和 97.61,将该参考基因组归类为 高质量基因组(表 S12)。这些结果共同表明,该参考基因组具有 高准确性、连贯性和一致性。
参考基因组的 Hi-C 热图 显示出 强烈的对角信号,并在不同染色体之间存在明显的 T2T(端粒到端粒)相互作用(图 2A,S3A,B)。此外,长末端重复序列(LTR)元件 主要集中在 染色体中央区域,而 基因主要位于染色体两端,二者的分布呈 负相关性(图 2B)。ONT 长读长序列比对到染色体上的覆盖度均匀(图 2B),进一步证明了 参考基因组的高质量。
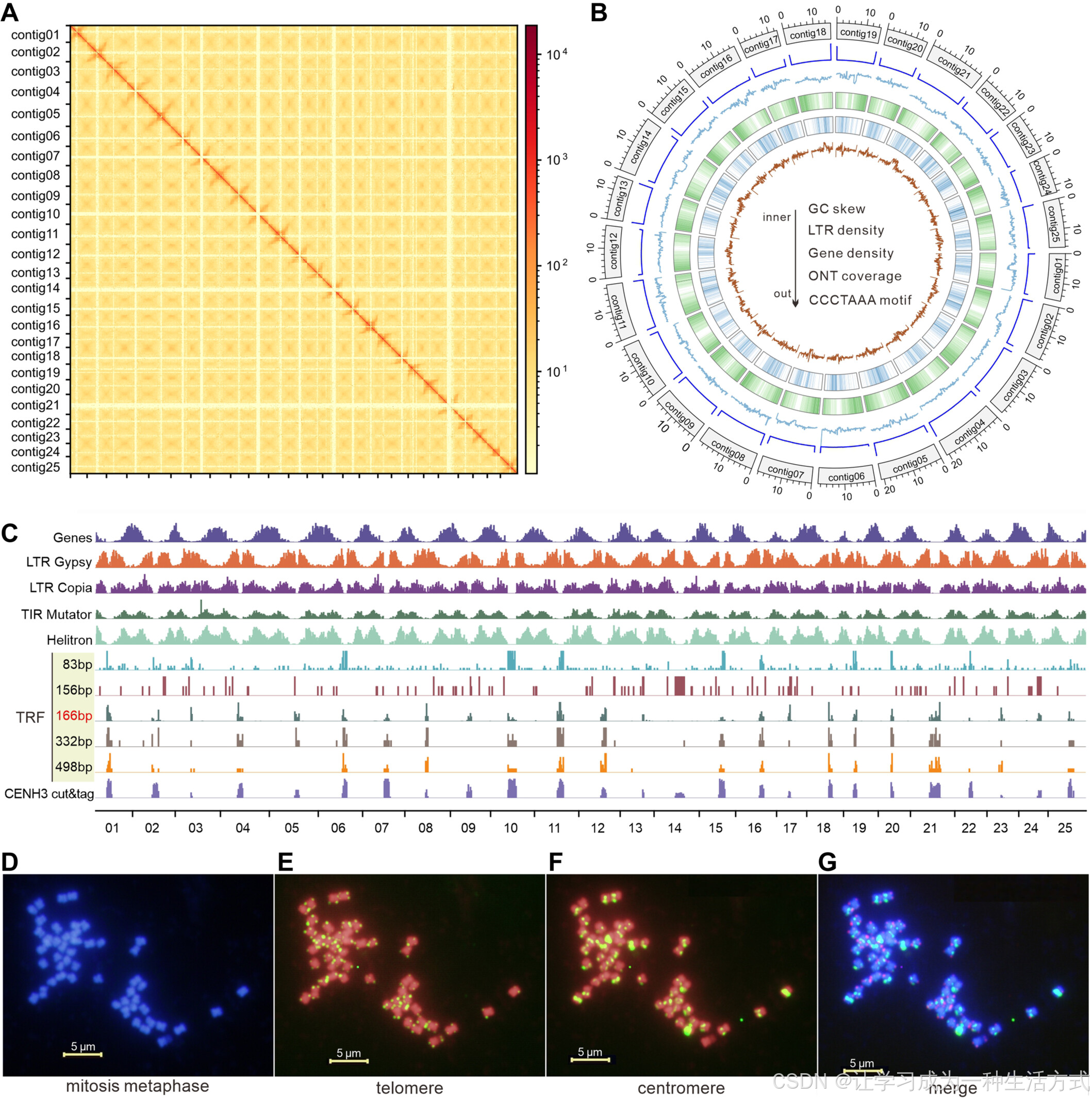
基因组组装及着丝粒和端粒的鉴定
(A) 参考基因组 25 条无缺口染色体的高通量染色体构象捕获(Hi-C)联系图。 (B) 参考基因组(Ref)的特征。从内至外的轨迹分别表示 GC 含量偏倚、长末端重复(LTR)密度、基因密度、Oxford Nanopore Technologies(ONT)覆盖度以及端粒重复序列 CCCTAAA 计数,窗口大小为 100 kb。 (C) 通过多种方法确定着丝粒位置,包括 基因和 LTR 分布分析、使用 Tandem Repeats Finder(TRF)软件识别串联重复序列,以及 CENH3 CUT&Tag 测序。 (D–G) 利用荧光原位杂交(FISH)技术在根部系统中验证端粒和着丝粒的位置。 (D) 在细胞有丝分裂中期观察到 50 条染色体。 (E) 端粒探针的绿色荧光信号出现在 50 条染色体的两端。 (F) 着丝粒探针的绿色荧光信号出现在 50 条染色体的中心。 (G) (E) 和 (F) 的合并结果,标尺为 5 μm。
为了识别着丝粒位置,我们使用 Tandem Repeats Finder(TRF) 软件检测长度在 50 至 500 bp 之间的串联重复序列。我们共鉴定出 456.7 kb 的串联重复序列,其中最常见的长度为 83、156、166、332 和 498 bp,分别占 2.5%、5.5%、47.6%、11.6% 和 8.2%,累计占比 75.5%(图 2C;表 S13)。166 bp、332 bp 和 498 bp 的串联重复序列沿染色体分布,主要集中在 低密度重复序列区域(如 Gypsy、Copia 和 Helitron)且远离富含基因的区域。这些高频串联重复区域可能为 染色体的着丝粒区域。TRF 分析表明 几乎所有 25 条染色体均检测到着丝粒区域,仅 contig13、contig14 和 contig22 信号较弱(图 2C)。
为了进一步确认着丝粒区域,我们使用 CENH3 CUT&Tag 测序 构建了测序文库。对显著峰区域的 motif 分析 发现 前 10 个 motif 与 TRF 识别的串联重复区分布高度一致。综合这些方法,我们确定了 所有 25 条染色体的着丝粒位置(图 2C)。着丝粒长度范围从 contig3 的 0.7 Mb 到 contig21 的 4.8 Mb,平均长度为 2.33 Mb(图 2D,S3C;表 S14)。
基于 端粒重复序列 CCCTAAA 设计了 荧光原位杂交(FISH) 探针,并在 YLL 菠萝品种的根尖细胞中进行实验。在有丝分裂中期,染色体两端均显示明显的绿色荧光(图 2D,E),证实了端粒的完整性。此外,结合 TRF 和 CENH3 结果,我们设计了针对着丝粒重复区域的探针,FISH 结果表明 着丝粒探针的绿色荧光信号明显强于端粒信号,这可能是由于 着丝粒重复序列长度更长且拷贝数更多(图 2G)。
综上所述,我们获得了高质量、无缺口、端粒到端粒(T2T)完整的菠萝参考基因组,并成功鉴定了完整的着丝粒。
获得高质量的菠萝基因结构注释
随着测序技术的进步以及组装软件的改进,越来越多的 T2T 基因组 得以构建(Li & Durbin, 2024)。然而,尽管基因组组装质量不断提高,基因结构注释的质量仍然存在不足(Salzberg, 2019)。在本研究中,我们利用 34 组转录组数据和同源蛋白数据,结合 MAKER 注释流程,对基因结构进行了优化。最终,共注释出 26,162 个结构基因,其 BUSCO 完整度评估得分为 94.5%(表 S5,S9)。
此外,我们利用团队自主开发的 IGV-GSAman 软件(IGV-GSAman Cookbook · 语雀),对基因结构注释进行了 人工优化。
基因结构注释错误主要可归为 六大类别:
-
非翻译区(UTRs)的错误注释
-
多个基因被错误地合并为一个基因座
-
未能注释出某些基因
-
部分基因结构未完整注释
-
基因结构的过度注释
-
单个基因被错误地拆分为多个基因(图 S4)。
例如,在 图 3A 中,初始基因注释 g02563.t1 跨越了两个区域,但覆盖度差异明显。在结合模式植物的蛋白质证据后,我们发现该区域实际上包含 两个不同的基因,分别为 lcfv2_02576.t1 和 lcfv2_02577.t1,推测它们可能具有不同的功能,并分别被注释为 “假设蛋白” (AT1G73470) 和 “雄性不育 1”(AT5G22260)。
在 图 3B 中,最初的基因结构注释 g01724.t1 和 g01725.t1 无法与 RNA-seq 比对数据的覆盖度和内含子边界对齐,表明原始注释存在错误。经过 人工修正,最终获得 优化后的基因结构 lcfv2_01727.t1。
在 图 3C 中,尽管 RNA-seq 读取数据质量较高,自动注释流程 未能识别出该区域的基因。通过人工校正,成功鉴定出 lcfv2_05233.t1,其编码序列的边界与 RNA-seq 数据中的内含子完全匹配,并被注释为 “阳离子氨基酸转运蛋白 2”(AT1G58030)。
在 图 3D 中,基因 g02657.t1 仅部分覆盖了 RNA-seq 读长数据。经过 手动校正,最终修正为 lcfv2_02671.t1,并被注释为 “参与基于肌动蛋白的叶绿体运动的驱动蛋白 2”(AT5G65460)。
综上所述,我们利用综合方法对菠萝基因组进行了高质量的结构注释,并通过人工优化提高了注释的准确性,为菠萝功能基因组学研究奠定了重要基础。
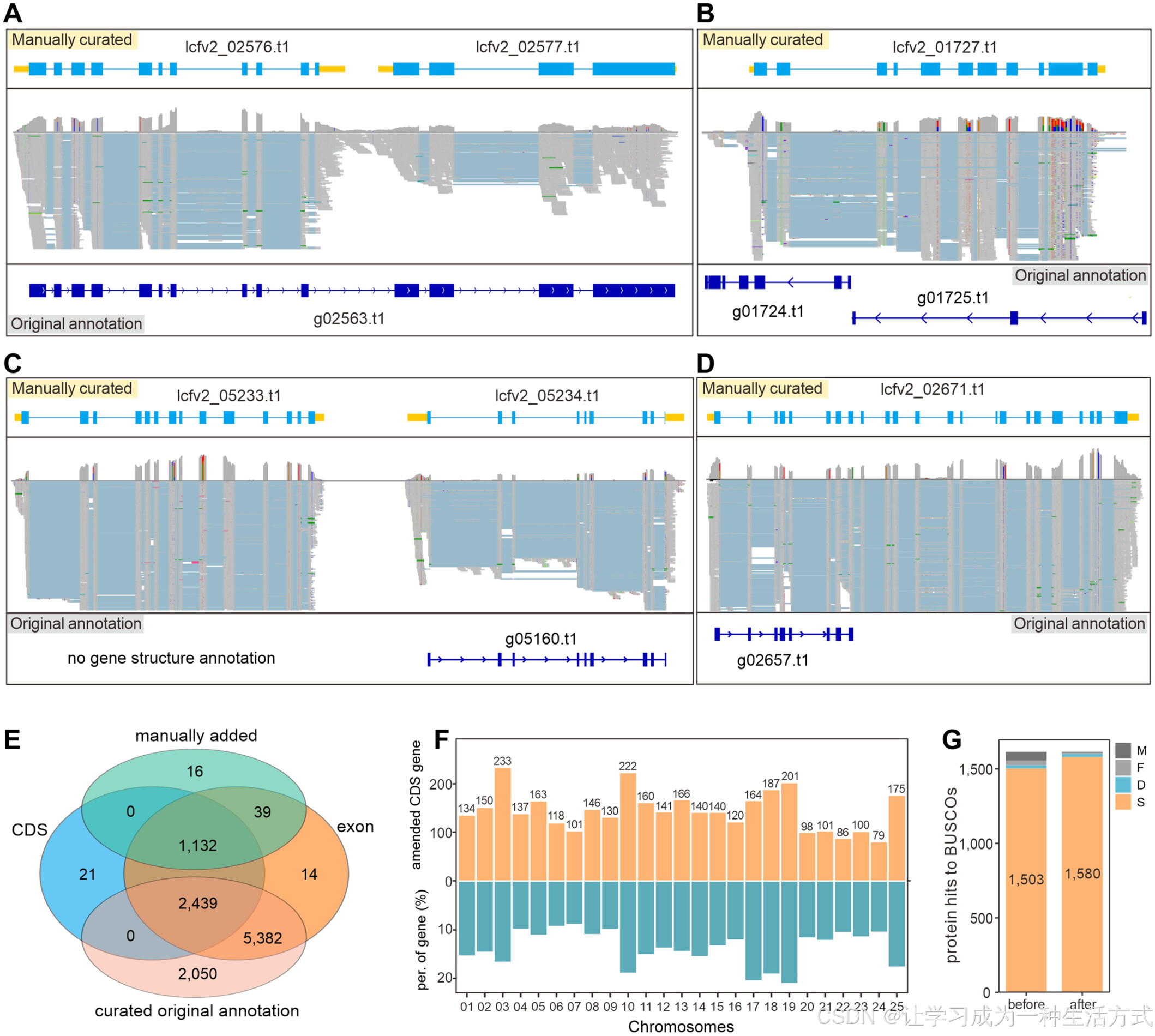
手动修正显著提升基因结构注释质量
(A–D) 基因结构注释中的四种错误类型:(A) 将多个基因错误地注释为一个位点;(B) 将单个基因错误地注释为多个基因;(C) 未注释的基因;(D) 基因结构部分注释错误。“原始注释”指的是从自动化流水线中获得的初始注释结果(详情见方法部分)。“手动修正”指的是经过手动校正后的注释。(E) Venn 图展示了手动修正的基因统计信息。CDS 和 exon 分别表示编码序列和外显子被修改的基因数量。(F) 每条染色体上发生蛋白序列变化的基因数量(上方条形图)和比例(下方条形图)。(G) BUSCO 评估基因结构注释的完整性,分别展示了修正前后的变化。S、D、F 和 M 分别对应单拷贝(single-copy)、重复拷贝(duplicated)、片段化(fragmented)和缺失(missing)的 BUSCOs。
在本研究中,我们对参考基因组中的所有基因进行了全面的手动修正,最终获得了 26,656 个结构基因。其中,共 11,058 个基因(占比 41.5%)经历了修正,其中 1,187 个基因(10.7%)为新增基因,而 9,871 个基因(89.3%)的注释得到了修正(图 3E)。此外,共有 3,592 个基因的编码序列(CDS)发生变化,占总基因数的 13.5%。每条染色体上被修正的基因数量从 79 到 233 不等,修正基因的比例介于 8.7%(contig07)到 21.0%(contig19)之间(图 3E, F;表 S15)。
为了验证基因结构修正的准确性,我们随机选取了 10 个修正后的基因模型进行聚合酶链式反应(PCR)验证。其中,7 个基因模型与修正后的结构一致(图 S5–S8)。其余 3 个基因未能成功克隆,可能是由于它们的时空表达模式在采样组织中不活跃。总体而言,手动修正显著提高了本研究中基因组注释的准确性。
在对修正前后的注释进行比较后,我们发现基于 embryophyta_odb10 数据集的单拷贝 BUSCO 数量增加了 77,从 1,503 提高到 1,580。此外,基因完整性指标从 94.5% 提高至 99.2%,表明基因结构注释质量得到了大幅提升(图 3G)。这一改进为菠萝功能基因组学研究奠定了坚实的基础。
与先前发布的基因组进行比较
迄今为止,共有六个菠萝基因组被测序:F153、MD2、CB5 (2019)、CB5 (2022)、Yugafu_3H 和 Yugafu_3C(Ming et al., 2015;Chen et al., 2019;Nashima et al., 2022)。这些基因组分别包含 4,553、8,654、1,867、479、1,763 和 1,804 个基因组缺口(表 1)。相比之下,本研究构建的参考基因组(以下简称“Ref”)为无缺口基因组,且其质量远优于以往的菠萝基因组。此外,Ref 基因组的大小为 423.8 Mb,与 F153 和 CB5,以及 Yugafu_3H 和 Yugafu_3C 之间的大小范围一致,表明其基因组大小符合该物种的典型范围(表 1)。
染色体比对显示,Ref 基因组与 F153、Yugafu_3H 和 Yugafu_3C 基因组呈现一对一的线性比对模式,这些基因组均基于遗传图谱进行锚定(图 S9)(Ming et al., 2015;Nashima et al., 2022)。与 F153 相比,Ref 在着丝粒区域(如 contig03、contig05 和 contig06)与 Yugafu_3H 和 Yugafu_3C 基因组的比对更好。这一发现表明,在染色体中央和近着丝粒区域,Ref 以及 Yugafu_3H 和 Yugafu_3C 的组装质量优于 F153。这种比对质量的提升主要归因于 Ref、Yugafu_3H 和 Yugafu_3C 采用了长读长测序数据进行组装(图 S9)。
与 CB5 (2019) 进行比对时,Ref 基因组显示出多个大规模结构重排现象,包括在 13 条染色体(如 chr2、chr3、chr7 和 chr8)上的易位和倒位。类似的结构变化在 CB5 (2022) 基因组中也有所观察,涉及 group1、group4 和 group6 等染色体组(图 S9)。值得注意的是,尽管 CB5 (2019) 和 CB5 (2022) 均属于鳞苞变种(bracteatus),但二者之间存在较多不一致性。此外,CB5 (2022) 的 Hi-C 热图显示,染色体上的端粒信号呈现无序和不规则的分布(图 S10)。然而,与现有的菠萝基因组相比,Ref 基因组几乎没有发生大规模的 DNA 片段倒位。结合 CB5 在进化关系上介于野生 YLL 和栽培品种 F153 之间的定位,我们推测 CB5 (2019) 和 CB5 (2022) 的组装质量仍有进一步优化的空间(图 S9)。
在本研究中,初始基因注释的 BUSCO 评估得分为 94.5%,手动修正后提升至 99.2%。这一结果不仅高于初始注释结果,也优于其他菠萝基因组:F153(88.0%)、MD2(82.5%)、CB5 (2019)(82.4%)、CB5 (2022)(84.0%)、Yugafu_3H(75.0%)和 Yugafu_3C(78.7%)(图 4A;表 1, S9)。值得注意的是,在这些基因组中,使用 Illumina 平台短读长测序组装的 F153 基因组,其基因结构注释质量最高,优于基于 PacBio 平台长读长测序组装的 CB5 (2019)、CB5 (2022)、Yugafu_3H 和 Yugafu_3C。这一发现表明,更完整的基因组组装并不一定意味着更优质的基因结构注释。
进一步分析发现,Ref 基因组的平均 CDS 长度最长,为 230.2 bp,其次是 MD2(222.2 bp)和 F153(217.5 bp)。相比之下,CB5 (2019) 的平均 CDS 长度仅为 186.9 bp,表明 CDS 长度与注释质量之间存在一定关联(表 S16)。
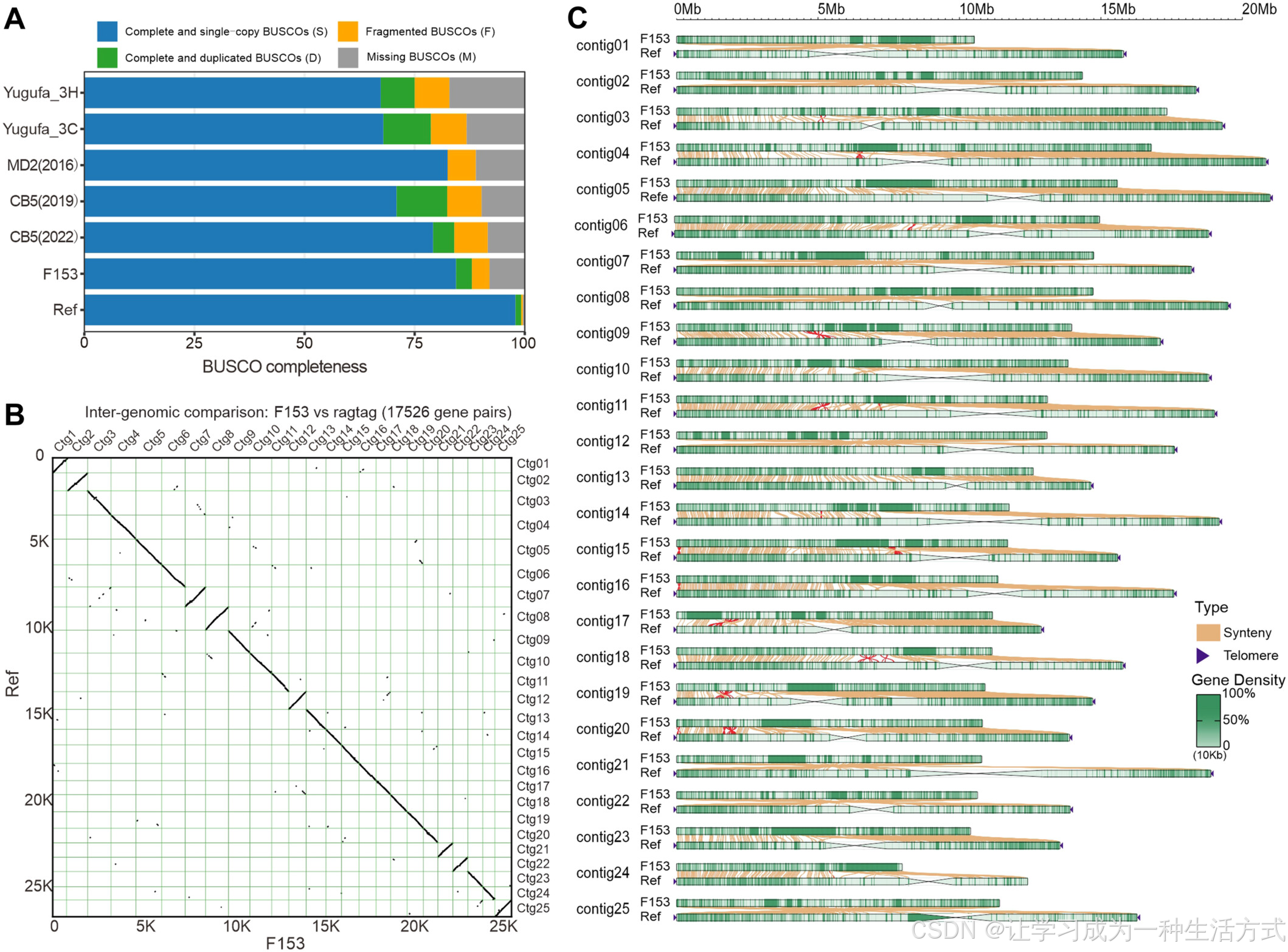
基因组序列和基因结构注释质量较高
(A) 本研究中的参考基因组(Ref)与六个已发表基因组(Yugafu_3H、Yugafu_3C、MD2 (2016)、CB5 (2019)、CB5 (2022) 和 F153)的 BUSCO 评估结果。(B) Ref 基因组与 F153 基因组的同源基因关系。(C) Ref 和 F153 之间的基因组共线性分析,其中红线表示染色体倒位区域。
我们进一步对 Ref 基因组与 F153 和 Yugafu_3H 进行了比较。基于编码序列的点阵图(dot-plot)分析表明,Ref 与 F153 之间具有很强的线性对应关系,表明两者在基因组水平上具有较高的相似性。相比之下,Ref 与 Yugafu_3H 之间的对应关系较弱,手动检查发现,这种差异主要是由于 Yugafu_3H 基因组的基因结构注释质量较低(图 4B, S11)。此外,在 Ref 与 F153 的比对中,尽管大多数区域表现出较好的共线性,但在 contig11、contig17 和 contig19 等染色体上仍检测到了 17 处较小的倒位(图 4C)。值得注意的是,在 F153 基因组中,与 Ref 基因组近着丝粒区域共线的区域通常具有较高的基因密度,尤其是在 contig14、contig19 和 contig23 上。这一现象表明 F153 在着丝粒区域的锚定质量相对较低(图 4C)。
Ref 基因组共包含 49 条相对完整的端粒,长度范围为 1.9 kb 至 24.4 kb,平均长度为 12.2 kb。唯一的例外是 contig24 末端未检测到端粒。在手动检查后,我们发现 F153 的染色体末端仅包含零碎的端粒重复序列(图 4C;表 S14)。
综上所述,本研究获得的参考基因组(Ref)在基因组组装和基因结构注释方面,均优于此前发表的六个菠萝基因组。
结构变异影响调控花青素合成的 MYB 基因表达
相比单核苷酸多态性(SNP),结构变异(SVs)对基因表达的影响更为显著(Alonge et al., 2020)。利用高质量的 Ref 基因组,我们在 BL、YLL 和 LY 菠萝品种中共鉴定出 7,209 处 SVs。其中包括 6,975 处缺失、54 处插入、75 处倒位和 105 处重复,总共覆盖 27.9 Mb。此外,共有 8,203 个基因(占基因总数的 30.8%)位于这些 SVs ±5 kb 的范围内(图 5A)。
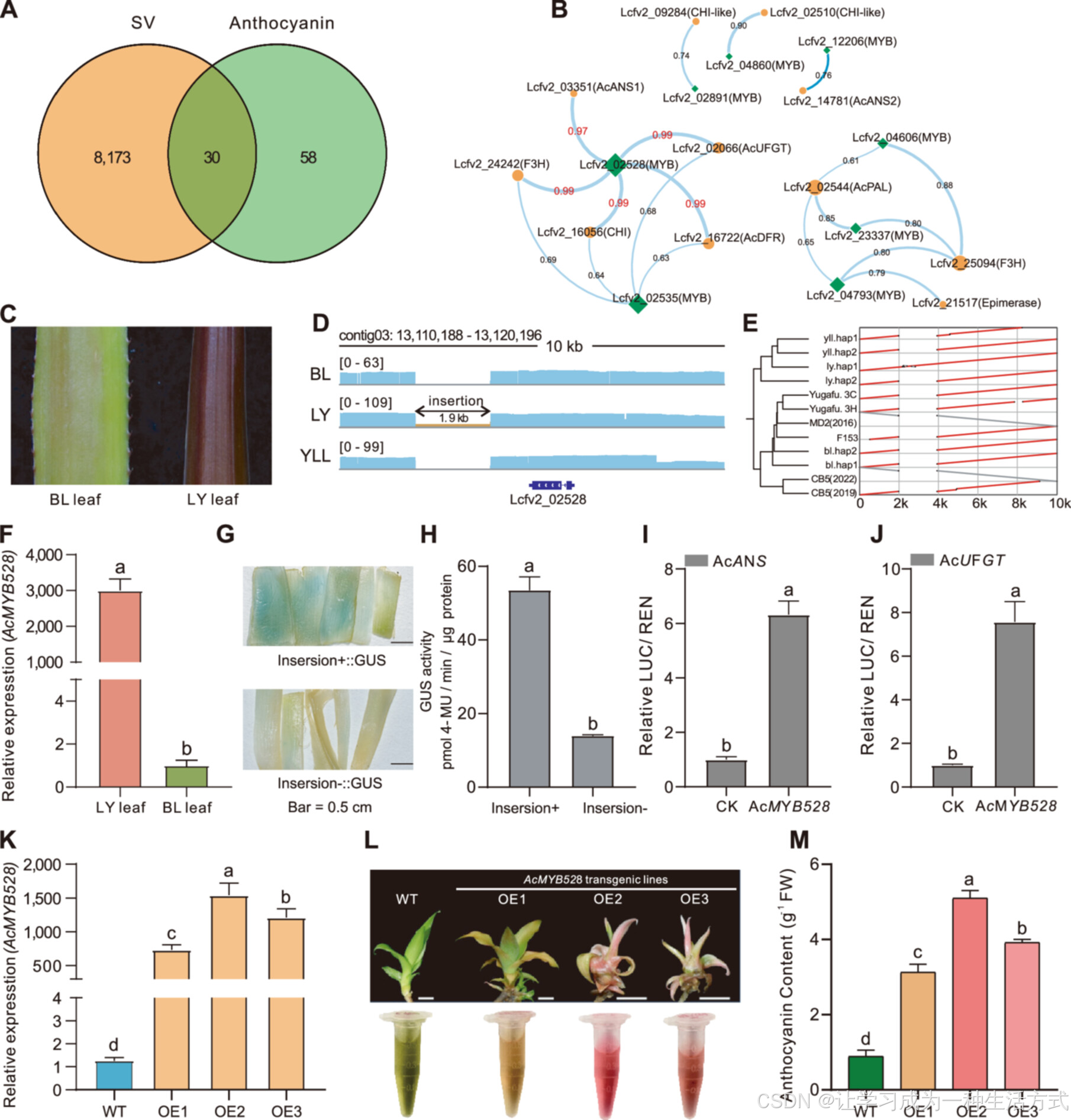
一种结构变异可能影响菠萝叶片花青素的生物合成
(A) Venn 图显示了花青素合成相关基因与位于 ±5 kb 结构变异区域的基因的重叠情况。(B) 30 个重叠基因的基因表达相关性网络,其中蓝色菱形代表 MYB 转录因子,黄色圆圈代表花青素合成的结构基因,连线表示 Pearson 相关系数,并标注相应的相关系数值。(C) LY(“Li Ye”)和 BL(“Ba Li”)品种的叶片外观。(D) 高保真(HiFi)测序数据表明,在 LY 中存在一个 1.9-kb 的杂合插入序列,而在 YLL(“Yu Ling Long”)和 BL 中则为纯合缺失。(E) 该 1.9-kb 插入序列仅存在于 LY.hap1,而在其他 11 个基因组中均未检测到。(F) 使用 qRT-PCR 定量检测 LY 和 BL 叶片中 AcMYB528 的表达水平。(G) 含有 1.9-kb 插入片段的调控区域(Insertion+::GUS)驱动 β-葡糖醛酸酶(GUS)瞬时表达,表现出更显著的 GUS 染色。(H) 带有 1.9-kb 插入片段的调控区域(Insertion+::GUS)相比于不含插入片段的调控区域(Insertion–::GUS)表现出更强的转录激活活性。(I–J) 双荧光素酶实验表明 AcMYB528 显著激活了结构基因(AcANS 和 AcUFGT)的表达。(K) 野生型(WT)和转基因植株中过量表达 AcMYB528 的相对表达水平。(L) WT 和转基因植株的表型,WT:野生型,OE:过表达株系,比例尺为 0.5 cm。(M) 过表达株系(OE)中的花青素含量显著升高,Duncan 多重范围检验用于显著性分析。
我们长期从事菠萝的食用和观赏品种育种工作,重点关注 LY 品种,该品种因植株呈现独特的红色而受到关注。在我们之前的研究中,通过鉴定同源蛋白,我们确定了 88 个与菠萝花青素合成相关的结构基因或 MYB 转录因子(Liu et al., 2017; Luan et al., 2023)。其中,有 30 个基因位于结构变异区域附近或与其重叠。结合此前报道的多阶段、多组织的菠萝基因表达数据(Mao et al., 2018)(图 S12),我们构建了一个相关性网络,以解析 MYB 转录因子与花青素合成结构基因之间的关系。研究发现,lcfv_02528 和 lcfv_02535 这两个 MYB 转录因子在花青素合成中发挥重要调控作用。其中,AcMYB528(lcfv_02528)与 AcCHI、AcDFR(双氢黄酮醇还原酶)、AcANS 和 AcUFGT(UDP-葡糖:黄酮 3-O-葡糖基转移酶)的相关系数均高于 0.97,表明它们之间存在强相关性。相比之下,lcfv_02535 与这些基因的最高相关系数仅为 0.69(图 5B)。
LY 品种的叶片颜色明显不同于 BL 和 YLL,其叶片呈深紫色,这是由于其高花青素含量(图 5C)。在 AcMYB528 基因下游,我们鉴定到一个 1.9-kb 插入序列,该插入在 LY 中为杂合状态,而在 BL 和 YLL 中为纯合缺失(图 5D)。在分析的所有菠萝基因组中,仅 LY 的单倍型 1(haplotype 1)包含该 1.9-kb 插入序列(图 5E)。同时,lcfv_02535 及其周围区域在三个品种中序列保持一致(图 S13)。基于以上发现,我们提出 AcMYB528(而非 lcfv_02535)是菠萝叶片呈红色的关键调控因子。
进一步检测叶组织基因表达水平发现,LY 叶片中的 AcMYB528 表达量显著高于 BL(图 5F)。这一高表达水平与 1.9-kb 插入片段的存在密切相关,该插入片段的调控区域表现出更强的转录激活能力(图 5G, H)。双荧光素酶实验进一步证实,AcMYB528 显著激活了花青素合成关键基因 AcCHS、AcDFR、AcANS 和 AcUFGT 的表达(图 5I, J, S14)。在转基因菠萝植株中过量表达 AcMYB528 的植株(OE1–OE3)中,该基因的表达量显著高于野生型(WT)(图 5K)。这一表达量的上升导致转基因植株叶片颜色明显加深,花青素含量显著增加(图 5L, M)。
菠萝基因组数据共享数据库的构建
目前,缺乏一个集中式的网络平台来共享菠萝基因组信息,这在一定程度上阻碍了菠萝育种和分子研究的进展。为了解决这一问题,我们建立了一个菠萝基因组数据库,网址为 https://ananas.watchbio.cn。该数据库整合了本研究获得的三个 T2T(端粒到端粒)菠萝品种基因组,以及其他公开可用的菠萝基因组数据。该数据库支持数据浏览、搜索、分析和下载(图 6A),为菠萝基因组研究提供了全面的资源平台。
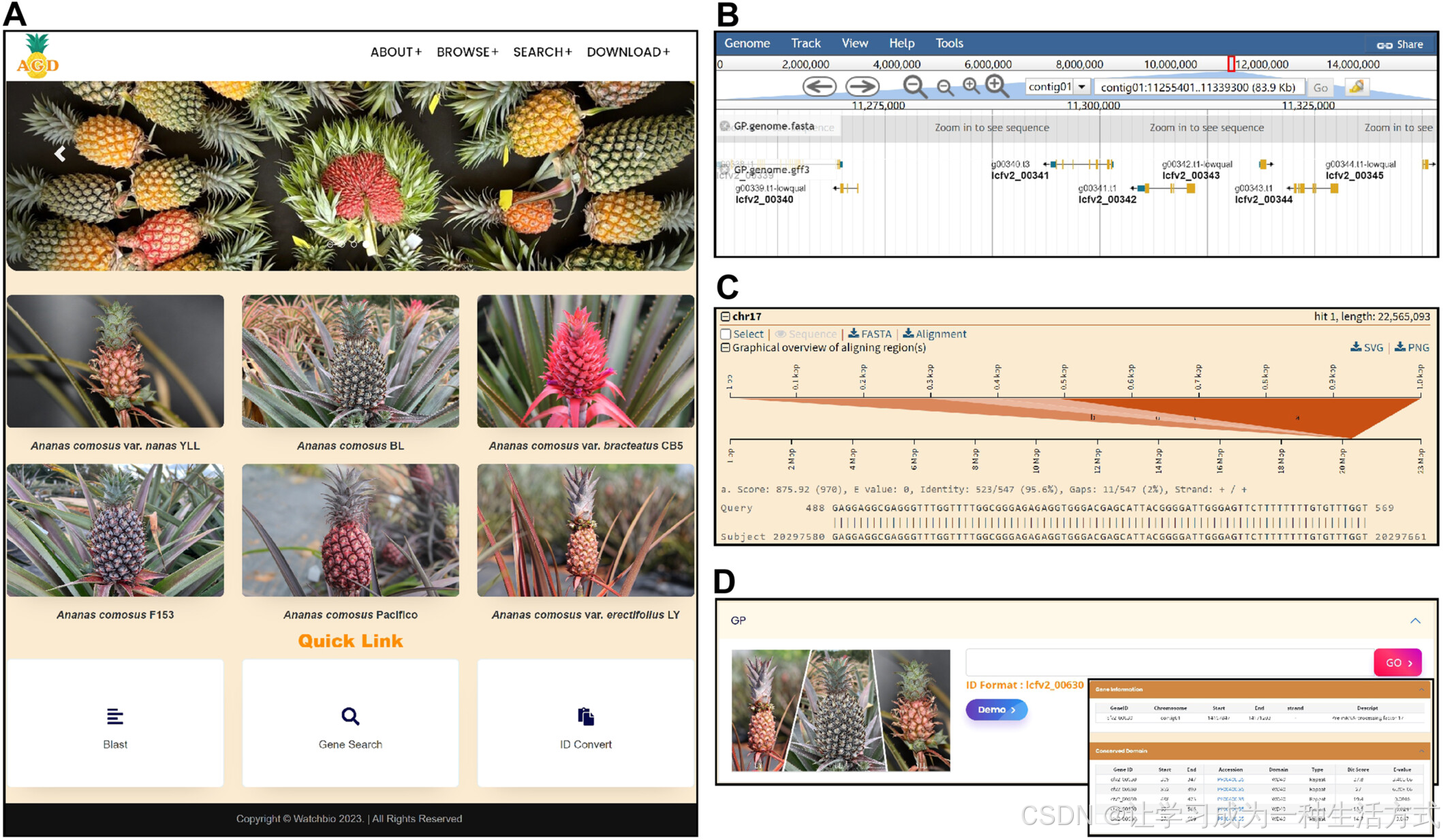
数据共享与数据库构建
(A) 菠萝基因组数据库的主页。 (B) 基于 Web 的基因组浏览器。 (C) 用于生物序列比对的基本局部比对搜索工具(BLAST)功能。 (D) 一个基因功能注释演示。
该数据库包含来自六个种质样本的基因组数据,代表了五种菠萝品种中的四种。其中,四个样本的单倍体基因组已完全解析。总体而言,该数据库包含 345,811 个基因、344,305 个转录本,以及 330,800 条基于序列比对的功能注释。此外,还提供了 288,797 条结构域注释,从而增强了该资源在基因组研究和育种计划中的实用性。
该数据库的 BROWSE 界面提供了不同菠萝样本的详细性状描述。用户可以通过在线基因组浏览器(图 6B)探索不同菠萝品种的染色体序列及基因结构注释。此外,为了提高菠萝基因组资源的可用性,我们开发了多个数据检索和分析接口,包括:(i) 用于生物序列比对的 BLAST 工具(图 6C);(ii) 基于基因 ID 或功能注释的数据检索和分析选项(图 6D);(iii) 快速 ID 映射工具,可用于关联菠萝基因 ID 与水稻或拟南芥基因组中的基因。此外,该数据库支持数据下载,使用户能够访问公开可用的序列数据,包括菠萝植物基因组、转录组、CDS 序列等。
讨论
端粒到端粒(T2T)菠萝基因组的构建为比较基因组学分析奠定了基础
凤梨属(Ananas comosus)包括五个品种。目前,仅 A. comosus var. bracteatus(CB5 (2019) 和 CB5 (2022))及 A. comosus var. comosus(F153、MD2、Yugafu_3H 和 Yugafu_3C)的基因组已被测序。这一有限的测序范围未能完整展现凤梨属的遗传多样性,从而阻碍了对菠萝进化和育种策略的深入研究。在本研究中,我们通过对两个额外品种——A. comosus var. erectifolius(LY)和 A. comosus var. microstachys(YLL)进行基因组测序,扩展了基因组数据。此外,我们还对皇后(Queen)品种 BL 进行了测序,该品种与 F153 在多个特征上存在差异,例如相对较长的货架期(Coppens d'Eeckenbrugge 等, 1997)。
对这些品种的基因组进行解码,极大地丰富了凤梨属的基因组资源。利用 HiFi 和 Hi-C 数据,我们成功组装了 LY、YLL 和 BL 的基因组。其中,大约 68%(17/25 条)染色体达到了端粒到端粒(T2T)水平。此外,我们构建了一个高质量的参考基因组 Ref,其中所有 24 条染色体均达到了 T2T 级别。仅 contig24 的短臂末端未能组装,可能是由于其近端端粒特性所致。其端粒距离染色体末端仅 2.66 Mb,这一情况与猕猴桃的观察结果一致(Yue 等, 2023)。Ref 基因组中的端粒长度在 3.3 kb 至 19.1 kb 之间,与水稻(5.1–10.8 kb)(Mizuno 等, 2006)和玉米(16–48 kb)(Chen 等, 2023a)中的端粒长度范围相当,表明该数据合理。初步鉴定的着丝粒大小范围为 0.7–4.88 Mb,类似于玉米(1.62–2.93 Mb)(Chen 等, 2023a)和西瓜(0.44–3.31 Mb)(Deng 等, 2022)。然而,确切的着丝粒大小仍需进一步实验验证。
此前已发表的菠萝基因组(F153、Yugafu_3C 和 Yugafu_3H)均基于遗传图谱搭建的框架组装,与 Ref 基因组表现出线性对应关系,表明不同菠萝品种之间的遗传变异较小(图 S9)。然而,Ref 与 CB5 之间的比较显示出大量的大片段倒位和易位。这一发现表明,早期的 SMRT 测序误差可能导致 CB5 基因组的 Hi-C 框架组装出现错误。未来,应用高保真(HiFi)测序技术可能有助于修正 CB5 基因组中的这些错误。尽管 F153、Yugafu_3C 和 Yugafu_3H 与 Ref 之间存在较强的线性关系,但这些基因组仍然存在多个未组装的端粒。此外,Yugafu_3C 中的 Aco3.3C04 染色体在组装过程中出现了冗余,而 F153 则缺少完整的端粒。这些观察结果进一步凸显了本研究构建的高质量菠萝参考基因组的卓越质量。
高质量菠萝基因结构注释为功能基因组研究提供参考
序列质量是任何广泛使用的参考基因组的基础,而准确的基因结构注释对于其广泛应用至关重要。随着时间推移,从 F153 到 CB5,再到 Yugafu_3C 和 Yugafu_3H,菠萝参考基因组的质量显著提高。然而,菠萝基因结构的注释仍然相对不足,这限制了基因功能研究和生物育种。在已发表的基因组中,F153 的基因结构注释质量最高,完整性评分达到 88.0%。本研究对高质量参考基因组进行了详尽的注释,依据 BUSCO 指标,注释完整性提高至 94.5%,较 F153 取得了显著提升。尽管取得了进展,但仍然存在相当数量的错误——在氨基酸序列中共检测到 3,592 处错误,占整个基因集的 13.5%,这还不包括 UTR 和外显子中的错误。为了解决这些问题,我们使用自主开发的基因结构注释校正工具 IGV-GSAman,对每条注释进行了系统修正。经过人工校正,我们的参考基因组注释完整性提升至 99.2%。这一改进趋势与其他物种基因组注释项目的观察结果一致。例如,Liu 等(2023)对黄花蒿基因组注释进行了修订,由于可变剪切和外显子结构的错误,他们修改了 8.1%(4,037/50,037) 的基因,使 BUSCO 完整性提高 1.1%(Liu et al., 2023)。类似地,在 CN14 桃基因组中,人工修正了 11.1%(3,981/35,826) 的转录本,使 BUSCO 评分从 98.98% 提升至 99.17%(Zhang et al., 2023)。这些数据表明基因结构注释错误普遍存在,即使是高度完整的基因组,如 98.98% 完整的 CN14 基因组,仍可能存在不准确之处(Zhang et al., 2023)。尽管本研究中的 Ref 参考基因组达到了 99.2% 的 BUSCO 完整性评分,但仍可能需要持续的后续修正。
结构变异分析揭示 MYB528 是菠萝叶色决定的候选基因
相比单核苷酸变异(SNVs),基因组结构变异(SVs)通常具有更大的影响,因为它们直接涉及基因的获得、缺失或融合(Scott et al., 2021)。在多种植物中,这些 SVs 与经济性状密切相关。例如,在玉米中,DRESH8 转座子的插入与穗径、穗长和穗重等产量决定性状相关(Sun et al., 2023)。最近的水稻研究表明,LTG1 启动子中的 SV 影响低温敏感性,而 GNP1 拷贝数的增加可正向调控籽粒数量(Wang et al., 2023)。在大豆中,基因 14G179600 的 SV 使不同种质材料之间的基因表达存在差异,可能影响黄化性状的形成(Liu et al., 2020)。此外,荔枝中含有一对 CO-like 基因的 3.7-kb 序列缺失,可能调控不同品种的果实成熟时间(Hu et al., 2022)。苹果中,MdMYB1 转录激活因子上游 UTR 的 LTR 反转座子插入,被发现是果实着色的关键因素(Zhang et al., 2019)。在本研究中,我们对菠萝的 SVs 进行了全面分析,并结合多组学数据,鉴定出调控菠萝花青素合成的关键基因 AcMYB528。实验验证表明 AcMYB528 与菠萝叶片着色相关,为叶色改良育种提供了新见解。进一步分析发现,AcMYB528 在其调控区存在一个 1.9-kb 的插入片段,这一插入导致 LY 品种中 AcMYB528 表达水平升高,从而促进叶片中花青素的积累。所有这些已鉴定的 SVs 可作为菠萝遗传和功能研究的参考,以及分子标记的潜在资源。
开发的菠萝基因组资源数据库为菠萝研究提供重要支持
在本研究之前,可用于菠萝研究的基因组资源极为有限。2005 年,Moyle 等人建立了一个菠萝生物信息学资源库,其中主要包含表达序列标签(EST)程序,用于组装剪接对齐序列(Moyle et al., 2005)。随后在 2018 年,Pineapple Genome Database(PGD)建成,用于共享与菠萝相关的基因组数据(Xu et al., 2018)。这些数据库的建立极大推动了菠萝分子生物学研究。然而,这两个数据库目前均已停止维护,且无法访问。此外,它们是基于单一菠萝样本构建的,无法有效满足现代基因组大数据研究的需求。为弥补这一缺陷,我们整合了本研究的数据,并结合公开可获取的菠萝基因组数据,建立了 Ananas Genome Database。该新数据库支持对四个品种的八种菠萝材料的基因组数据进行探索、分析和下载,为菠萝研究团队提供了强有力的支持。
结论
本研究首次对野生菠萝品种的基因组进行深入解析,构建了高质量的菠萝参考基因组,并完成了全面的基因注释。通过广泛的结构变异分析和 RNA 测序数据整合,鉴定出调控菠萝叶色的关键基因 MYB528。此外,为促进菠萝基因组数据的共享,本研究建立了全面的菠萝基因组数据库,提升了菠萝基因组研究的可访问性和协作能力。
材料与方法
样本采集
三种菠萝品种(BL、LY 和 YLL)在中国海南 国家热带植物种质资源库菠萝分库 进行种植,地理位置为 北纬 19°29′17″,东经 109°29′4″。该地区海拔 130 m,属于 热带季风气候,年均气温 23°C,相对湿度 85%,年降水量 1,500 mm。所有采集的样本立即在液氮中冷冻,并储存于 −80°C 直至进一步使用。
HiFi 文库构建与测序
从 BL、LY 和 YLL 菠萝品种的叶片中提取基因组 DNA,采用 DNeasy Plant Mini Kit(QIAGEN,德国) 进行 DNA 提取,并使用 Agilent 4200 Bioanalyzer(Agilent Technologies,美国) 评估 DNA 完整性。约 15 μg 基因组 DNA 通过 g-Tubes(Covaris,美国) 进行片段化,并使用 AMPure PB 磁珠 纯化。随后,利用 SMRTbell Express Template Prep Kit 2.0(Pacific Biosciences,美国) 构建 SMRT bell 文库,并在 BluePippin 系统 上进行片段筛选,以分离 约 11 kb 长度的分子。然后,使用 DNA/Polymerase Binding Kit(Pacific Biosciences,美国) 对 SMRT bell 模板进行引物退火并结合聚合酶。最终,在 Pacific Biosciences Sequel II 平台 进行测序,测序工作由 Annoroad Gene Technology 公司 完成。
Hi-C 文库构建与测序
Hi-C 文库的构建基于 BL、LY 和 YLL 菠萝品种的幼叶,测序工作由 Annoroad Gene Technology 公司(北京,中国) 进行,采用的方法参考 Xie 等(2015)。文库构建完成后,在 Illumina HiSeq 平台 上进行 150-bp 双端测序(paired-end sequencing)。
Nanopore 文库构建与测序
从 BL、LY 和 YLL 菠萝品种的新鲜叶片中提取高质量基因组 DNA,采用 十六烷基三甲基溴化铵(CTAB)法 进行提取。对于 Oxford Nanopore 超长测序,文库采用 SQK-LSK110 连接文库构建试剂盒(Oxford Nanopore Technologies,英国) 按标准协议进行构建。纯化后的文库加载到 R9.4.1 Spot-On Flow Cells 进行测序,并使用 PromethION 测序仪(Oxford Nanopore Technologies,英国) 进行 72 小时测序,测序工作由 Annoroad Gene Technology 公司 完成。
CUT&Tag 文库构建与测序
从 BL、LY 和 YLL 菠萝品种的叶片中提取细胞核,并将其固定于 磁珠 上,然后孵育于 一抗 CENH3(Abcam, ab72001, 英国) 和 pG-Mnase 酶(Vazyme,南京,中国) 溶液中。随后,在 pG-MNase 作用 10 min 后回收基因组 DNA 片段。回收的 DNA 片段经过纯化后扩增为文库,并进行测序。更详细的方法请参考 Wei 等(2021)。
表型数据收集与分析
根据 国际植物新品种保护联盟(UPOV) 发布的 《菠萝(Ananas comosus (L.) Merr.)特异性、一致性和稳定性测试指南》(www.upov.int),测量,-vo0l042s/) BL、LY 和 YLL 菠萝品种的 株高、冠幅、果重、果实横径、果眼数目及可溶性固形物含量。测量数据采用 双尾 t 检验 进行统计显著性分析。
基因组组装
Ref 基因组组装流程如 图 S2 所示。简要来说,三种菠萝品种(BL、LY 和 YLL)的 HiFi 和 Hi-C 读取数据 经过 hifiasm(Cheng et al., 2021) 进行单倍型分辨组装,最终获得 六个单倍型(haplotypes)。
在组装过程中,携带 端粒重复序列(CCCTAAA) 的单倍型被识别为 完整的 T2T 单倍型(telomere-to-telomere haplotypes),随后使用 Mummer(Kurtz et al., 2004) 比对到 F153、CB5 和 Yugafu 基因组。最终,人工筛选 最长的同源染色体序列 构建 Ref 参考基因组。
在此基础上,以 Ref 基因组 作为参考,将 六个单倍型锚定到染色体水平。此外,ONT 读取数据 通过 NECAT 组装工具(Chen et al., 2021) 进行组装,并用于填补已分相单倍型组装中的 缺口(gaps),最终获得 完整的单倍型序列。
基因组注释与人工校正
基因模型注释
基因模型的注释采用 MAKER 注释流程(Campbell et al., 2014),该流程整合了 AUGUSTUS 和 SNAP 进行 ab initio 预测,并结合同源证据进行优化。同源证据包括 SwissProt(Bairoch 和 Apweiler, 1999) 的蛋白序列、菠萝基因组 F153、CB5 和 Yugafu,以及通过 Trinity(Grabherr et al., 2011) 从 RNA-seq 数据 de novo 组装 的转录组序列。为了提高预测准确性,使用 MAKER 进行了三轮迭代注释。随后,基因模型经过 三轮 PASA(Haas et al., 2003) 更新,最终得到 BUSCO 完整度 94.5% 的初始注释。
人工校正
为了提高注释质量,我们使用 自研软件 IGV-GSAman(IGV-GSAman Cookbook · 语雀) 进行人工校正。RNA-seq 读取数据通过 STAR(Dobin et al., 2013) 映射到参考基因组,生成 BAM 文件。此外,利用 拟南芥和水稻的同源蛋白数据 通过 miniprot(Li, 2023) 进行预测建模。两个数据集共同导入 IGV-GSAman,对初始注释进行手动修正。
着丝粒定位
结合三种方法进行分析:
-
LTR 元素注释:使用 Extensive De novo TE Annotator(Su et al., 2021) 进行 LTR 元素注释,参数设置为 “–sensitive 1”。
-
串联重复序列识别:使用 TRF(Benson, 1999),仅保留长度 >50 bp 的重复序列。
-
CUT&Tag 分析:
-
CUT&Tag 读取数据通过 BWA(Li et al., 2009) 映射到相应单倍型。
-
使用 macs2(Zhang et al., 2008) 识别信号峰(阈值设定为 1e−5)。
-
参考基因组的信号峰序列基于六个单倍型的映射关系进行提取。
-
使用 STREME(Bailey, 2021) 进行基序分析,统计 前 10 种显著富集的基序 并绘制分布图。
-
荧光原位杂交(FISH)
荧光原位杂交实验参照 Su et al.(2019) 进行。
-
选取 YLL 菠萝根尖细胞 作为实验材料。
-
设计 两种荧光探针,分别针对菠萝的潜在 端粒重复序列(CCCTAAA, 7 nt) 和 着丝粒重复单元(166 nt)。
-
探针使用 荧光染料 标记,染色体核型分析基于重复序列进行。
-
通过 共聚焦显微镜 采集图像。
高通量染色质构象捕获(Hi-C)热图
-
Hi-C 数据的 正向和反向测序读取 通过 BWA mem(v. 0.7.17) 映射到参考基因组,参数设置为 “-A1 -B4 -E50 -L0”(Li et al., 2009)。
-
采用 HiCExplorer(v. 2.1.1)(Ramírez et al., 2018) 计算 Hi-C 接触矩阵并绘制热图。
Circos 图绘制
-
统计基因组 GC 偏斜、CCCTAAA 元素计数。
-
从 基因模型 GFF 文件、LTR 注释文件、ONT 覆盖 BAM 文件 获取 基因密度、LTR 密度、ONT 覆盖度。
-
采用 100-kb 窗口 计算基因组特征,并使用 TBtools-II(Chen et al., 2023b) 可视化 Circos 图。
基因组与基因注释完整性评估
-
采用 BUSCO(v. 5.4.2),基于数据库 embryophyta_odb10(Manni et al., 2021) 评估基因组和基因模型的完整性。
遗传距离树构建
-
选取 四个已发表基因组(F153、CB5、MD2、Yugafu_3C、Yugafu_3H)、本研究的 六个单倍型 及 外群物种,采用 jolytree(Criscuolo, 2020) 构建遗传距离树,参数默认。
基因组比对分析
-
采用 nucmer(Marçais et al., 2018) 比对参考基因组与 F153、CB5、Yugafu_3C、Yugafu_3H,参数设置为 “-l 200 -c 500 -g 1000”。
-
过滤 delta 文件,参数为 “-i 90 -l 200 -o 90 -1”。
-
使用 mummerplot(Kurtz et al., 2004) 可视化基因组比对结果。
染色体示意图
-
整合 LTR、端粒、着丝粒 数据,使用 RIdeogram(Hao et al., 2020) 在 R 中绘制染色体示意图。
点图比对(Dot-plot)
-
从 F153、CB5、Yugafu_3C、Yugafu_3H 和 Ref 提取蛋白序列,使用 TBtools-II(Chen et al., 2023b) 处理。
-
采用 JCVI(v. 0.8.4)(Tang et al., 2015) 进行可视化,参数设定为 “–cscore = 0.99”。
共线性分析(Synteny analysis)
-
采用 GenomeSyn(v. 1.2)(Zhou et al., 2022) 进行 F153 基因组与参考基因组的共线性比对,并绘制共线性图谱。
结构变异(SV)鉴定
-
HiFi 读取数据(BL、LY 和 YLL)映射至参考基因组,使用 ngmlr(Sedlazeck et al., 2018) 进行比对。
-
结果文件经 SAMtools(Li et al., 2009) 排序和索引。
-
Sniffles(Sedlazeck et al., 2018) 进行 SV 识别。
-
SURVIVOR(Jeffares et al., 2017) 合并多样本 SV 数据,并再次调用 Sniffles 生成最终变异文件。
-
筛除未知基因型 SV 及长度 <50 bp 或 >100 kb 的 SV。
-
保留 7,209 个 SV,包括 缺失(DEL)、插入(INS)、倒位(INV)和重复(DUP)。
-
使用 Bedtools(Quinlan, 2014) 确定 SV 位点 ±5 kb 范围内的基因。
转录组分析
我们基于先前研究(Mao et al., 2018)共使用了来自 14 个组织的 39 个 RNA-seq 文库。转录组数据经过 Trimmomatic(Bolger et al., 2014)进行质量剪切,并使用 STAR(Dobin et al., 2013)比对至参考基因组。最终,使用 StringTie(Pertea et al., 2016)计算每百万映射片段的每千碱基转录片段值(FPKM)。
相关系数网络构建
计算与花青素生物合成相关的 MYB 转录因子和结构基因的表达水平,并在 R 语言环境下计算皮尔逊相关系数。去除相关系数低于 0.6 的数据后,使用 Cytoscape(Otasek et al., 2019)进行网络可视化。
植物材料
在遗传实验中,菠萝品种 Shen Wan(A. comosus cv. Shen Wan,简称 SW)的愈伤组织被用作农杆菌介导的稳定遗传转化受体材料。烟草(Nicotiana benthamiana)被用于双荧光素酶实验。
总 RNA 提取及 RT-qPCR 分析
总 RNA 由康为世纪生物公司 RNA 提取试剂盒(CW2598S,北京,中国)提取。反转录-定量 PCR(RT-qPCR)实验使用反转录试剂盒(Yeasen, 11141ES60, 上海,中国)和定量试剂(11203ES08, 上海,中国),并在 BIO-RAD 荧光 qPCR 仪器(Bio-Rad/CFX Touch)上进行。RT-qPCR 结果以 AcActin 为内参基因,并使用 2−ΔΔCT 方法计算基因相对表达量。实验引物列于表 S17,所有实验均进行三次生物重复,并以误差线表示。
基因克隆与表达载体构建
根据本研究参考基因组设计正义和反义引物(表 S17)。cDNA 从 LY 叶片中克隆,扩增片段连接至 T 载体 pCloon 007,并经 Sanger 测序验证后用于后续实验。使用 Gateway 技术(Karimi et al., 2002)构建过表达载体(表 S17)。AcMYB528 目的片段经 BP 重组反应连接至 pDONR221 载体,重组质粒 pDONR-AcMYB528 再经 LR 重组反应连接至 pK7WG2D 过表达载体,该载体由 CaMV 35S 启动子驱动。最终构建的质粒经农杆菌 Agrobacterium tumefaciens GV3101 转化至菠萝愈伤组织,转化方法参考 He et al.(2023)。
双荧光素酶报告实验
AcCHS、AcDFR、AcANS 和 AcUFGT 的启动子片段分别克隆至 p0800-Luc 载体的 KpnI 和 SalI 位点(Hellens et al., 2005)。成功构建的 p0800-Luc 载体随后转化至 A. tumefaciens GV3101。将 pK7WG2D-AcMYB528 质粒与启动子载体按 1:5 体积比混合,并注射至 N. benthamiana 幼叶进行瞬时共转化表达分析。使用双荧光素酶报告系统(E1910, Promega, Wisconsin, Madison, USA)测定萤火虫荧光素酶与海肾荧光素酶的活性比值,并按照生产商说明进行实验。
AcMYB528 邻近调控变异的活性分析
根据 LY 基因组序列设计调控区扩增引物。以 LY DNA 为模板,扩增出两个版本的序列:一个含 1.9 kb 插入序列,另一个不含该插入序列(图 S15)。获得的序列克隆至表达载体 pBI121,并替换其中的 CaMV 35S 启动子,构建 1.9 kb 插入驱动(Insertion+::GUS)和无插入驱动(Insertion−::GUS)两种 β-葡糖苷酶(GUS)表达载体。参考 Zhang et al.(2024)的方法,在菠萝基部叶片中分析这两种序列的活性。
统计分析
所有实验重复至少三次,结果以标准差(SD)表示。数据采用 t 检验分析,当 P < 0.05 时,结果被认为具有统计学显著性,显著性水平以不同字母标注。


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









