







Frame2seq: structure-conditioned masked language modeling for protein sequence design
https://github.com/dakpinaroglu/Frame2seq
Abstract
机器学习彻底改变了计算蛋白质设计,使蛋白质主链生成和序列设计取得了重大进展。 对于蛋白质序列设计,编码器-解码器模型已达到最先进的精度,本研究介绍了一种用于蛋白质序列设计的结构条件掩码语言模型,与自回归方法相比,可以一次性生成序列。
模型在CATH4.2测试集上性能由于ProteinMPNN,实现了49.1%的序列恢复率,且推理速度提高了6倍以上,为了探究Frame2seq生成超出类自然蛋白质序列的空间的新颖设计能力,文中测试了26个Frame2seq设计的与起始序列同一性较低的de nove 主链。
Introduction
蛋白质是驱动所有生命系统中的细胞过程的分子,因此设计出具有新功能的蛋白质的能力在生物技术和医学中具有广泛的应用。传统,计算蛋白质设计依赖于基于物理原理和模拟。最近基于机器学习的方法取得了有希望的进展。
计算蛋白质设计在各种应用中取得了很高的实验成功率,包括生成新的蛋白质折叠和对称寡聚物、蛋白质-蛋白质结合物设计和基序支架。 本质上,所有这些方法首先生成蛋白质主链,然后进行固定主链序列设计作为第二步。
Methods
Datasets
使用CATH4.2数据中,长度不超过500的单链蛋白。训练集、验证集和测试集中分别有18024、608和1120个没有拓扑重叠的链(拓扑重叠的链指的是在蛋白质的三维结构中,两个或多个肽链彼此交错或缠绕在一起,形成一种拓扑学上的复杂结构)。这样划分是为了结构多样性被聚类,并允许在许多折叠中测试泛化性。为了进行直接的比较,我们对在相同的数据集上训练的模型进行基准测试。
Structure-conditioned masked language model
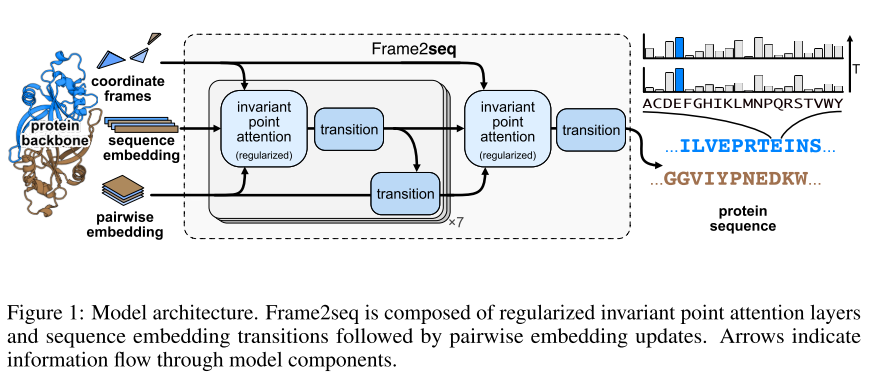
Frame2seq是一个平移和旋转不变的编码器模型,它将蛋白质骨架作为输入并生成蛋白质序列,在一次推理中,模型可以设计一个完整的蛋白质序列。模型使用invariant point attention(IPA)来保持不变性。模型从输入骨架的坐标计算旋转和平移,构建AlphaFold2所述的坐标框架。
为了获取序列嵌入,我们计算二面角(phi、psi和Omega扭转角)以及绝对位置嵌入。
成对嵌入:径向基的残基间的距离和残基对之间的相对位置索引。坐标系、序列嵌入、成对嵌入经过节点和边更新操作。然后是最终节点更新并转换到序列维度。 IPA 层和过渡层用于节点更新。 我们通过将成对嵌入和更新的序列嵌入传递到 MLP 的两层来更新边。
IPA的注意力正则化
假设在训练过程中引入可容忍的难度可以带来更好的模型性能。 为了增加训练难度而不停止模型最小化分类交叉熵损失的能力,文中探索了 IPA 的特征丢失和注意力正则化。 我们通过在训练期间随机屏蔽残基对之间的注意力权重来实现正则化 IPA 层(算法 1)。 发现 20% 的掩蔽率是最佳的,可以提高模型性能(表 S2)。 下图为IPA的伪代码其中9为本文所改进的随机掩码。
Result
Frame2seq从结构中恢复自然序列
文中与在CATH4.2数据上训练的proteinMpnn模型进行基准测试,模型的天然序列的恢复率为49.1%,比ProteinMPNN好2%。使用所有 5 个 AlphaFold2 模型(其中 3 次循环且无模板)预测了天然序列、ProteinMPNN 设计和 Frame2seq 设计的结构。我们计算了有和没有 MSA 的 AlphaFold2 高置信率 (%) 和 AlphaFold2 成功率 (%)。 我们发现 ProteinMPNN 和 Frame2seq 具有相似的高置信度和成功率。对测试数据集目标进行平均时,Frame2seq 推理速度比 ProteinMPNN 快约 6.2 倍。
固定主干设计方法的不正确残基预测揭示了它们对某些氨基酸类型的潜在组成偏差。成分偏差的计算方法是从真值中减去预测的残基类型出现次数,再除以总数。我们计算了所有氨基酸类型的ProteinMPNN和Frame2seq的组成偏差,发现Frame2seq的偏差总体较小(图2A)。
接下来,我们研究了 ProteinMPNN 和 Frame2seq 的性能如何取决于残基埋藏度。为了衡量残基埋藏度,我们计算了 8 个最近残基的平均原子 Cβ 距离(埃)(核心残基较低,表面残基较高)。我们发现,残基埋藏对两种模型的影响相似,即核心残基比表面残基更容易恢复。与 ProteinMPNN 相比,Frame2seq 的原生序列恢复率提高了 2.0%,这主要体现在目标物的表面残基上(图 S1)。
‘burial埋藏度是指残基相对于蛋白质表面的位置’
核心残基受到更多的周围残基的限制和约束,所以其结构和位置相对更稳定,更容易被模型准确捕捉。而表面残基则受到更多的外部影响,其结构和位置可能更加灵活和多变,因此更难被模型准确预测。
Frame2seq准确估计了自己预测的误差


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









