






WGCNA 简明指南|3.使用WGCNA实现网络可视化
WGCNA 系列
WGCNA的基础教程到此就告一段落,之后将以已发表文章内的示例进行实战讲解。需要基础教程的全部代码及示例数据的朋友可以
点赞+在看,并转发朋友圈集赞10个或者赞赏10元。
WGCNA 系列
参考
数据准备
在R中可视化网络
可视化基因网络
eigengenes网络可视化
将网络数据导出到网络可视化软件
导出到Cytoscape
往期
参考
本文主要参考官方指南Tutorials for WGCNA R package (ucla.edu),详细内容可参阅官方文档。
其它资料:
WGCNA - 文集 - 简书 (jianshu.com)
WGCNA分析,简单全面的最新教程 - 简书 (jianshu.com)
WGCNA:(加权共表达网络分析)_bioprogrammer-CSDN博客
WGCNA如何从module中挖掘关键基因_庐州月光的博客-CSDN博客
数据准备
在R中可视化网络
可视化基因网络
# 模块检测时的计算,重新算一次
dissTOM = 1-TOMsimilarityFromExpr(datExpr, power = 6);
# 对dissTOM进行power转换,使中等强度的连接在热图中更加明显
plotTOM = dissTOM^7;
# 设置对角线为NA以得到更好的图
diag(plotTOM) = NA;
# 绘图
sizeGrWindow(9,9)
TOMplot(plotTOM, geneTree, moduleColors, main = "Network heatmap plot, all genes")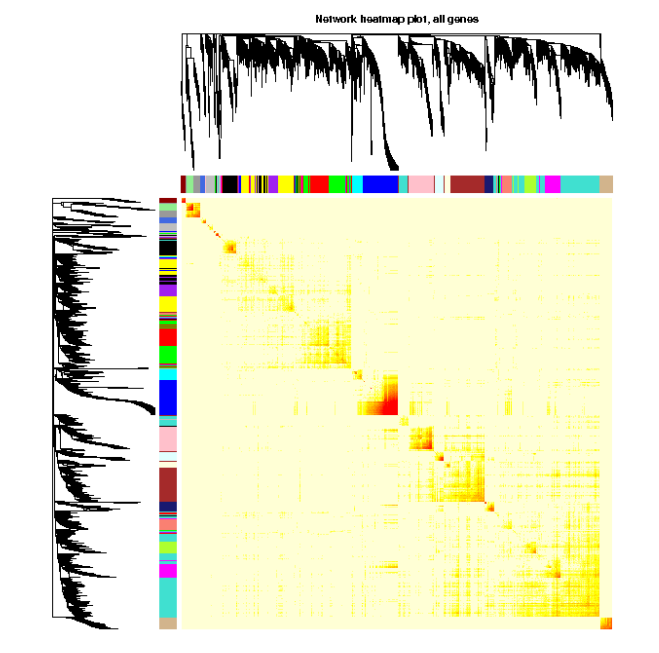
部分基因可视化TOM矩阵
全部基因生成热图可能需要大量的时间。可以限制基因的数量来加快绘图速度。然而,一个基因子集的基因树状图通常看起来不同于所有基因的基因树状图。在下面的例子中,将绘制的基因数量限制在400个。
nSelect = 400
# 为了可重复,设置随机数种子
set.seed(10);
select = sample(nGenes, size = nSelect);
selectTOM = dissTOM[select, select];
# 没有简单的方法将聚类树限制在基因的一个子集,所以我们必须重新聚类
selectTree = hclust(as.dist(selectTOM), method = "average")
selectColors = moduleColors[select];
# 绘制
sizeGrWindow(9,9)
plotDiss = selectTOM^7;
diag(plotDiss) = NA;
TOMplot(plotDiss, selectTree, selectColors, main = "Network heatmap plot, selected genes")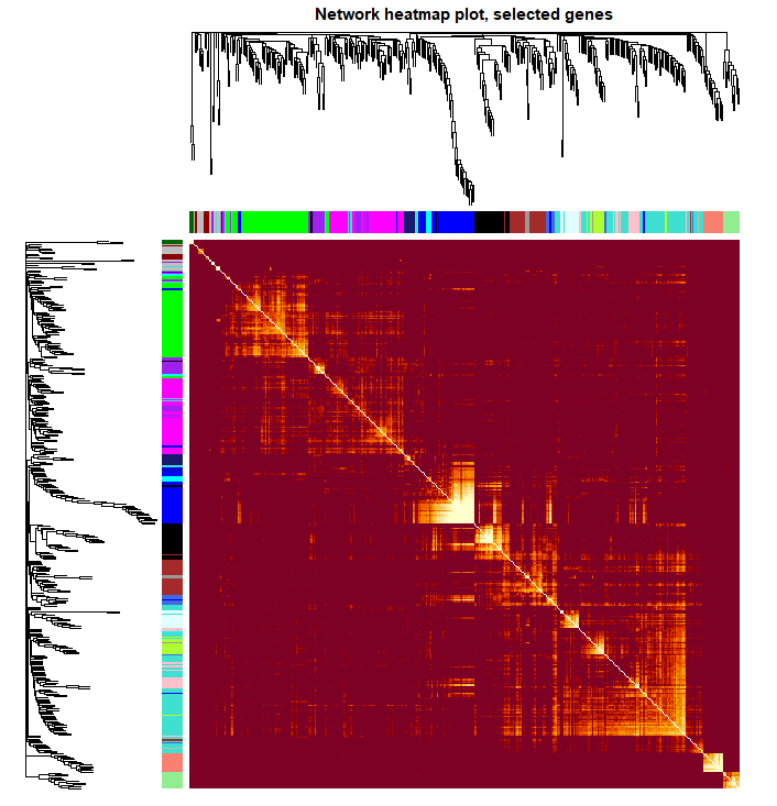
eigengenes网络可视化
# 重新计算模块 eigengenes
MEs = moduleEigengenes(datExpr, moduleColors)$eigengenes
# 提取临床特征weight
weight = as.data.frame(datTraits$weight_g);
names(weight) = "weight"
# 在eigengenes模块中加入临床特征weight
MET = orderMEs(cbind(MEs, weight))
# 绘制eigengenes和临床特征weight之间的关系图
sizeGrWindow(5,7.5);
par(cex = 0.9)
plotEigengeneNetworks(MET, "",
marDendro = c(0,4,1,2),
marHeatmap = c(3,4,1,2),
cex.lab = 0.8, xLabelsAngle= 90)
# 分别绘制
# 绘制树状图
sizeGrWindow(6,6);
par(cex = 1.0)
plotEigengeneNetworks(MET, "Eigengene dendrogram", marDendro = c(0,4,2,0),
plotHeatmaps = FALSE)
# 绘制热图
par(cex = 1.0)
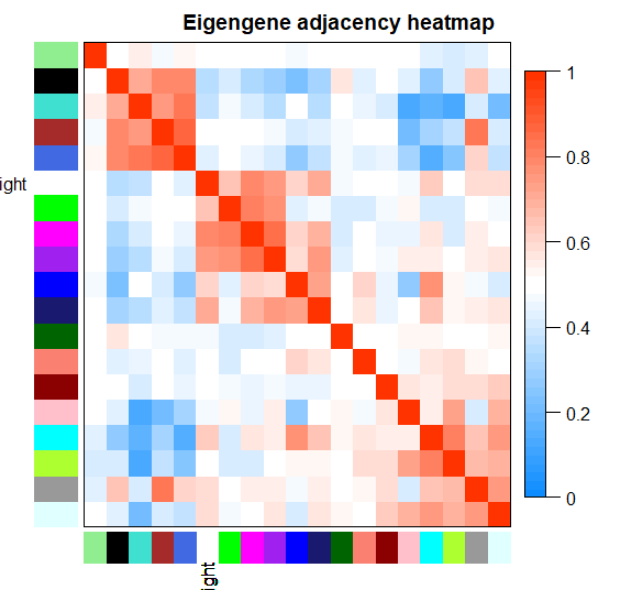
plotEigengeneNetworks(MET, "Eigengene adjacency heatmap", marHeatmap = c(3,4,2,2),
plotDendrograms = FALSE, xLabelsAngle = 90)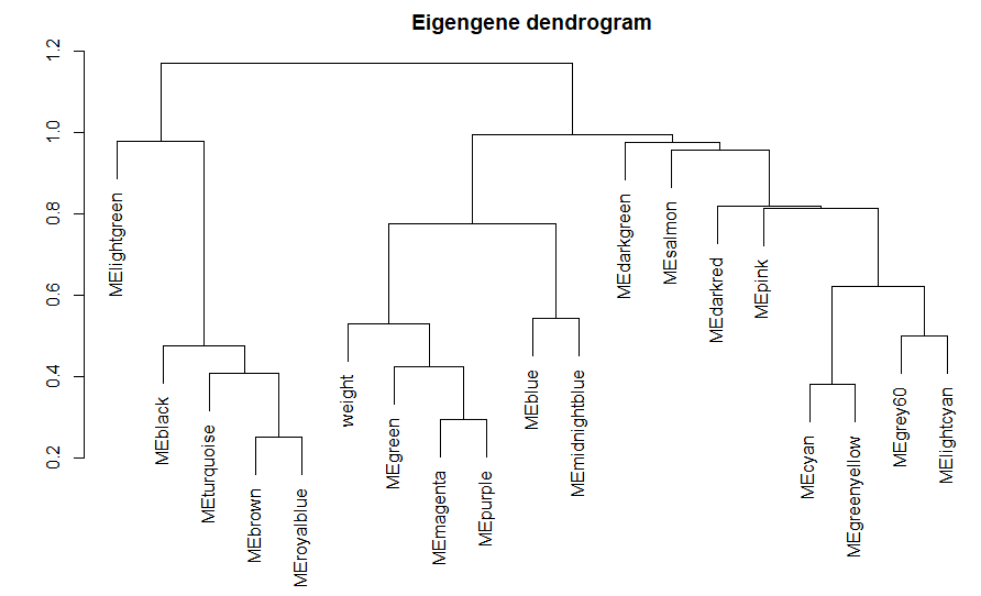
将网络数据导出到网络可视化软件
导出到Cytoscape
# Recalculate topological overlap if needed
TOM = TOMsimilarityFromExpr(datExpr, power = 6);
# Read in the annotation file
annot = read.csv(file = "GeneAnnotation.csv");
# 以红色和棕色模块为例
modules = c("brown", "red");
# Select module probes
probes = names(datExpr)
inModule = is.finite(match(moduleColors, modules));
modProbes = probes[inModule];
modGenes = annot$gene_symbol[match(modProbes, annot$substanceBXH)];
# Select the corresponding Topological Overlap
modTOM = TOM[inModule, inModule];
dimnames(modTOM) = list(modProbes, modProbes)
# Export the network into edge and node list files Cytoscape can read
cyt = exportNetworkToCytoscape(modTOM,
edgeFile = paste("CytoscapeInput-edges-", paste(modules, collapse="-"), ".txt", sep=""),
nodeFile = paste("CytoscapeInput-nodes-", paste(modules, collapse="-"), ".txt", sep=""),
weighted = TRUE,
threshold = 0.02,
nodeNames = modProbes,
altNodeNames = modGenes,
nodeAttr = moduleColors[inModule]);cyt中有edge和node数据,可以导入cytoscape进行可视化。

















 3895
3895

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









