







文献介绍

「文献题目」 SpaceX: gene co-expression network estimation for spatial transcriptomics
「研究团队」 Veerabhadran Baladandayuthapani(美国密歇根大学)、Xiang Zhou(美国密歇根大学)
「发表时间」 2022-09-30
「发表期刊」 Bioinformatics
「影响因子」 6.9
「DOI」 10.1093/bioinformatics/btac645
摘要
空间解析转录组的分析能够了解细胞环境和转录调控之间的空间相互作用。特别是,与标准差异单基因分析相反,对组织中不同空间位置或细胞类型的基因-基因共表达的表征能够描绘空间共调控模式。为了增强空间转录组学技术推动生物发现的能力和潜力,作者开发了一个统计框架来检测由细胞类别或组织域形式的不同类群组成的空间结构组织中的基因共表达模式。
作者开发了 SpaceX(空间相关基因共表达网络),这是一种贝叶斯方法,用于识别跨基因的共享和类群特定的共表达网络。SpaceX 使用过度分散的空间泊松模型和高维因子模型,该模型基于降维技术以提高计算效率。作者通过模拟展示了通过考虑(增加)空间相关性和适当的噪声分布来提高共表达网络估计和结构的准确性。利用 SpaceX 对小鼠下丘脑和人类乳腺癌的两个空间转录组数据集进行深入分析,检测到下丘脑数据中与认知能力相关的多个中心基因以及乳腺癌肿瘤区域中的多个癌症基因(例如胶原蛋白家族)数据。
SpaceX R 包可在https://github.com/bayesrx/SpaceX上获取。
1 前言
空间转录组学的最新技术进步促进了生物组织中高通量 RNA 测序数据的获取,同时也考虑了空间信息。为了破译组织内的空间细胞结构,空间转录组技术,例如 10× Genomics Visium 和 Slide-seq,使用空间索引条形码和 RNA 测序来进行定量分析转录组与单个组织切片中的空间信息。这些新技术可以帮助理解许多生物系统的空间组织,包括发育中的脑组织和肿瘤微环境,并有助于表征细胞环境和基因表达之间的空间相互作用并描绘健康组织和患病组织之间的组织组织差异。空间转录组学的一个主要兴趣点是研究组织中细胞间信号传导的空间变化,这可能是疾病病因学以及心理或行为模式的基础。
转录组分析的一个重要方面侧重于基因共表达模式,因为基因往往通过生物网络自然地相互连接。基于网络的模型提供了一个简单且可解释的框架来表征各种生物系统中复杂的基因相互作用模式。基因共表达网络通常使用基于图的表示来表征,其中节点代表基因,边描述基因之间的关联或调节相互作用。已经开发了几种网络方法来检测基因共表达网络并识别基因调控群落或模块,以便产生可能与潜在的生物和调控途径相关的生物学见解,了解因果组织或细胞类型并可能影响疾病风险和结果。与仅测量单个基因表达修饰的标准差异表达分析相比,识别病例和对照等条件之间网络结构的变化可以揭示特定疾病的重要补充信息。
在标准单细胞研究中构建基因共表达网络的大多数现有计算方法本质上涉及降维步骤,该步骤实现两个目标:一是避免维度灾难并提高计算可行性;其次是在减少噪声的同时保留固有维度。然而,现有的网络方法没有纳入空间转录组学中至关重要的空间信息。仅提出了有限数量的工作来研究空间转录组学中的基因相互作用或共表达模式。具体来说,Salamon et al. (2018) 最近的一项工作提供了空间共表达网络的可视化、基因图卷积神经网络方法 (Yuan and Bar-Joseph, 2020) 和 Giotto (Dries et al., 2021) 方法,特别关注配体和受体的相互作用。此外,所有这些方法都假设给定样本中有一个共同的基因网络。然而,人们可能不会期望通用网络能够捕获所有空间依赖性,因为基因组特征可能表现出样本内基于区域特异性异质性的特定空间位置。例如,这些区域可以是病理学上不同的区域(例如癌症中的肿瘤与正常区域)或基于不同的细胞类型,因此这些区域可以表现出截然不同的共表达模式。这就激发了对基于网络的模型的需求,该模型考虑了空间信息以及一个基本假设,即存在整个空间中常见的共享(全局)共表达网络以及不同空间中的局部变化网络区域。
为此,作者提出:空间依赖基因共表达(spatially dependent gene co-expression, SpaceX)网络模型,用于推断具有共享和区域特定组件的空间转录组数据的基因共表达网络。Figure 1 显示了 SpaceX pipeline 的整体概念流程。给定组织切片的图像(Fig. 1A)用于分析覆盖在组织切片上的空间基因表达,并在空间位置上有(已知)聚类注释(Fig. 1B)。所得的基因表达矩阵数据矩阵以及组织上每个空间位置的空间定位和聚类注释信息(Fig. 1C)作为 SpaceX 模型的输入。SpaceX 使用过度分散的空间泊松模型与高维因子模型(Panel H)配对来推断共享和类群特定的共表达网络(Fig. 1D and E)。最后,这些网络用于下游网络分析,以检测跨空间区域的基因模块和中心基因(Fig. 1F and G),以进行生物学解释。

(A)一张感兴趣区域的组织切片图像。(B)借助单细胞测序技术,从该组织切片中记录空间基因表达和生物标志物。(C)获得了
个 genes 和
个 locations 的基因表达矩阵,其中 locations 被分为
个 clusters。作者将 SpaceX 模型应用在基因表达矩阵上以获得共享的共表达网络(D)和 cluster-specific 共表达网络(E)然后进行 hub gene 分析和社区检测。对于所有网络图,node 大小和 edge 宽度分别与连接基因的数量和基因共表达水平成正比。最后,作者在 shared 和 cluster-specific 网络中检测生物保守的 communities(F)和 hub genes(G)。组织切片(A)和空间基因表达(B) 图片由 Loupe Browser 改编自 10× genomics 网站。(H)SpaceX 算法详细工作流程。
简而言之,SpaceX 采用贝叶斯模型,通过在确定网络拓扑时结合空间信息来推断空间变化的共表达网络。概率模型(在第 2 节中进一步详细介绍)能够量化不确定性,并基于相干降维技术来提高计算效率。通过严格的模拟(第 3 节),作者证明 SpaceX 模型能够准确地恢复网络结构并提高不同空间相关结构的估计精度。作者将 SpaceX 模型应用于小鼠大脑成像和乳腺癌数据集,以确定区域特定网络(第 4 节)。进一步的下游分析检测到基因模块和相关 hub genes 的多个群落。SpaceX 能够从小鼠下丘脑数据中识别出与行为模式和认知能力相关的多个基因。类似地,作者从乳腺癌的肿瘤区域检测到多种胶原蛋白和癌症特异性基因。
2 SpaceX 模型
2.1 方法概述
就输入数据结构而言,表示观察到的基因表达数据 genes,以及空间索引 clusters, 大小为 。这些 clusters 可以是不同细胞类型的细胞类型特异性注释,也可以是注释不同空间域的空间连续的 clusters。作者构建了一个 维网络,其中 基因之间的依赖关系可以通过具有一组 vertices 和一组 edges 的无向图来描述。两个 nodes 之间的 edge (E) 表示它们之间的共表达水平,它是使用相似性度量定义的,在本文的例子中是相关系数。在 SpaceX 模型中,作者构建的网络由以下两个层次组件组成:
-
一个‘共享’组件,代表跨空间域基因之间的全局共表达网络; -
一个‘cluster’特定组件,代表给定(第 c 个)cluster 的本地或 cluster-特定基因共表达网络。
这种分解实现了两个目标。首先,它能够精确描述跨空间 clusters 保守和修改的共表达网络组件,从而实现更连贯的解释。其次,这有利于降维技术,使整个方法可针对大型网络进行扩展。如 Figure 1H 中的图形工作流程所示,SpaceX 算法将基因表达矩阵、空间位置和 cluster 注释作为输入。第一步,算法使用泊松混合模型估计潜在基因表达水平,同时调整协变量和空间定位信息。在下一步中,它利用潜在基因表达的稀疏层次因子模型来获得共享和 cluster 特定的共表达网络。接下来,第 2.2 节将讨论该模型的详细构建,随后将在第 2.3 节中讨论该模型的联合估计和实施。
2.2 模型构建
根据上述目标,可以从使用多种空间转录组学技术收集的基因表达数据中推断出基因共表达网络(10× Genomics)。基因表达数据通常以 counts 的形式收集,代表在单个细胞中成像的给定转录本的条形码 mRNAs 的数量或映射到空间位置上给定基因的测序 reads 的数量。表达计数测量值随空间位置的不同而变化,其空间坐标在实验期间记录在 个空间区域中。使用 表示在 空间位置的第 个 cluster 的第 个 gene 的表达。使用泊松对数线性框架以 counts 的形式直接对基因表达数据进行建模。根据之前的研究,count 数据过度分散,每个基因的方差高于平均值。因此,作者引入了一个随机效应项来考虑简单泊松模型无法解释的额外变异性。
为此,作者将基因表达数据建模为:
其中 是第 个 cluster 第 个位置处第 个基因的未知(空间)速率参数, 是归一化因子。作者认为 cluster 特异性的所有基因的 counts 总和是 。 是一个 矩阵,表示第 个 cluster 中所有基因和空间位置的速率参数。cluster 特异性和空间相关的速率参数 之后使用加法对数线性方程建模,即
模型(2)中五个术语的具体上下文解释如下:(i) 是一个 矩阵包含系数的基因特定向量,包括第 c 个cluster 的截距。这里, 表示对应于第 c 个 cluster 的第 g 个基因和第 l 个协变量的系数。 是第 c 个 cluster 的协变量矩阵。可能的协变量可以包括 cluster 特定 batch size、细胞周期信息或与实验相关的任何其他协变量。在加性模型(2)中, 解释了第 c 个 cluster 特定协变量效应。(ii) 考虑了 cluster 特定的空间效应,其中每行 使用均值为 0 的多元正态分布和空间解析高斯核作为协方差进行建模,即 。这里 考虑了 cluster 特定的空间相关性,它允许灵活地对 cluster 内空间相关性的异构程度进行建模。(iii) 是由共享载荷矩阵 和共享因子矩阵 组成的共享结构。(iv) 类似地, 是 cluster 特定结构,其中 是 cluster 特定的载荷矩阵并且 是 cluster 特定的潜在因子。(v) 最后, 是一个 cluster 特定的特殊误差矩阵,这意味着每个元素都遵循一个正态分布。
在模型(2)公式中,作者有效地利用降维技术来确保基因共表达网络的可扩展构建。作者的方法基于潜在因子模型,该模型利用低维结构,特别是对于多视图数据,同时识别共享共表达网络并单个 cluster 特定网络。通过因子模型和协方差矩阵之间的对应关系,这使我们能够推断出基因共表达网络的两个重要且分层的组成部分:
-
'Shared' 组件表示为 这是共享因子和 的第 个元素的协方差矩阵,表示共享结构中第 i 个基因和第 j 个基因之间的共表达。
-
类似地,'Cluster' 特定基因共表达水平由下式表示 , 其中 是 cluster 特定因子的协方差矩阵。第 个元素表示第 i 个基因和第 j 个基因之间的 cluster 特异性共表达。
2.3 贝叶斯估计算法
为了拟合模型 (2),作者使用易于处理的贝叶斯估计程序以及计算高效且可扩展的算法,如下所述。与计算密集型的 full-scale Markov chain Monte Carlo 算法相反,作者将整个模型估计解耦为两个关键部分(i)空间泊松混合模型和(ii)层次因子分析模型,并且这两个组件在算法中按顺序链接:
-
空间泊松混合模型 (sPMM) 是一种加法结构,连接对数尺度 具有协变量效应、空间效应和剩余的基因特异性效应。作者拟合 sPMM 并将无法解释的变异性和潜在基因表达转移到层次因子分析模型中。
-
然后使用层次因子分析模型推断共享和 cluster 特定的基因共表达网络。
对于估计过程,作者使用 PQLseq 算法,这是一种可扩展的惩罚准似然算法,用于具有高斯先验的 sPMM 来获取潜在基因表达。为了拟合层次因子分析模型,遵循 Vito 等人的算法,作者将乘法伽玛收缩先验置于共享和 cluster 特定载荷矩阵之前,即 和 。共享和 cluster 特定的潜在因子(分别为 和 )是通过 Vito 等人中描述的方法从头自动选择的。关于方法的估计程序和方法论新颖方面的更多详细信息在 Supplementary Section SA 中提供。
2.4 共表达网络构建与推理
共表达网络的构建:使用 SpaceX 算法,获得共享的( )和 cluster 特定的( )后验样本的协方差。后验均值估计 和 用于构建共表达网络,分别如 Figure 1D and E 所示。这些协方差被转换为相关矩阵以进行推理和解释。例如,共享相关矩阵( )的第 (i,j) 个元素 表示第 i 个基因和第 j 个基因之间的共享相关性。
共表达网络的推论总结:共表达网络中两个基因之间的显着边定义为 ,其中 是基于错误发现率 (FDR) 的截止值,如 Baladandayuthapani 和 Ni 等人所做的那样。还可以为特定于 cluster 的网络提供类似的解释。如果第 i 个基因和第 j 个基因在所有 cluster 中存在共表达,则认为这两个基因之间的边是保守的。在实际数据分析中,检测 top edges 并讨论跨 cluster 保守的 edges(Section 4)。作者使用共表达网络进行下游分析,通过优化网络结构中分区的模块化来检测 hub genes(Fig.1G)和具有基因模块的社区(Fig.1F)。根据与每个基因相连的边数来检测来自共享网络和特定 cluster 网络的 hub genes。如果某个特定基因是跨 cluster 的 hub gene,称该 hub gene 在生物学上是保守的。
3 模拟研究
作者在一系列空间依赖性下模拟真实数据应用程序(Section 4),评估 SpaceX 模型在合成数据集中的性能。作者的核心假设是,通过考虑空间相关性,随着空间相关性的连续增加,应该能够实现更好的估计和共表达网络恢复(共享的和特定于 cluster 的)。
模拟设计:作者考虑 个 clusters 的空间位置,cluster 大小为 。作者将共享因子加载的维度设置为 ,并表示 cluster 特定加载维度 。为了模仿单细胞 RNA 谱的稀疏和零膨胀性质,作者在共享和通用载荷矩阵的列上随机分配零,即 和 并根据 分布生成其余元素。cluster 特定设置以三元组形式提供 : (700, 5, 70), (500, 4, 75), (300, 3, 67), (1000, 6, 55), (1700, 7, 60), (200, 2, 65), (600, 3, 75),其中第一个对应于第一个 cluster,后面是其余 6 个 clusters 的设置。对于总共 N = 5000 个位置中的每一个,作者使用 SpaceX 模型模拟了 G = 160 个基因的表达水平 (2)。
作者考虑三个级别的空间依赖性:(i)high( ,表示为 ),(ii)medium( ,表示为 ,(iii)low( ,表示为 )空间相关性。基于真实的数据探索, 、 、 所有细胞类型相应的诱导空间相关性分别为 0.88,0.80,0.61,分别在给定距离 0.01 处;额外细节和空间相关衰减图在 Supplementary Section SB.1 提供。作为基线比较器,作者采用两种非空间设置:(iv)在泊松混合模型中完全忽略空间信息(用 表示),即每一行 遵循均值为 0 且协方差矩阵恒等的多元高斯分布,并且(v)不考虑 PMM 和空间信息。此设置表示为 。所有模拟结果均来自 50 多个重复数据集。
共表达估计:作为共表达估计准确性的度量,作者使用 RV 系数来量化真实协方差(共表达)矩阵与估计协方差(共表达)矩阵之间的相似性,RV 值接近 1(0) 意味着更高(更低)的相似度。在 Figure 2A 中,显示了共享( )和 cluster 特定的( )先前提出的五个设置的协方差矩阵的 RV 系数的箱线图。可以看出,空间设置比非空间设置更好地估计共表达值 RV 系数。在空间设置中,共表达水平的估计精度随着空间相关性水平的增加而增加。例如,对于共享网络,方法(v)-(i)通过 RV 系数测得的中值估计精度分别为 0.83、0.88、0.91、0.92、0.96。空间和非空间设置之间的成对 t 检验显示网络估计精度显着提高(P-value < 0.05)。作者观察到不同范数度量(欧几里德、对数欧几里德、根欧几里德、黎曼)的相似模式,并且在 Supplementary Section SB.2 中给出了此类详细讨论。共享和 cluster 特定负载的性能准确性( 和 )在 Supplementary Section SB.2 (Fig. SB.3 and B.4) 中进行了描述。总而言之,作者观察到估计精度随着空间信息的结合和更高水平的诱导空间依赖性而增加。
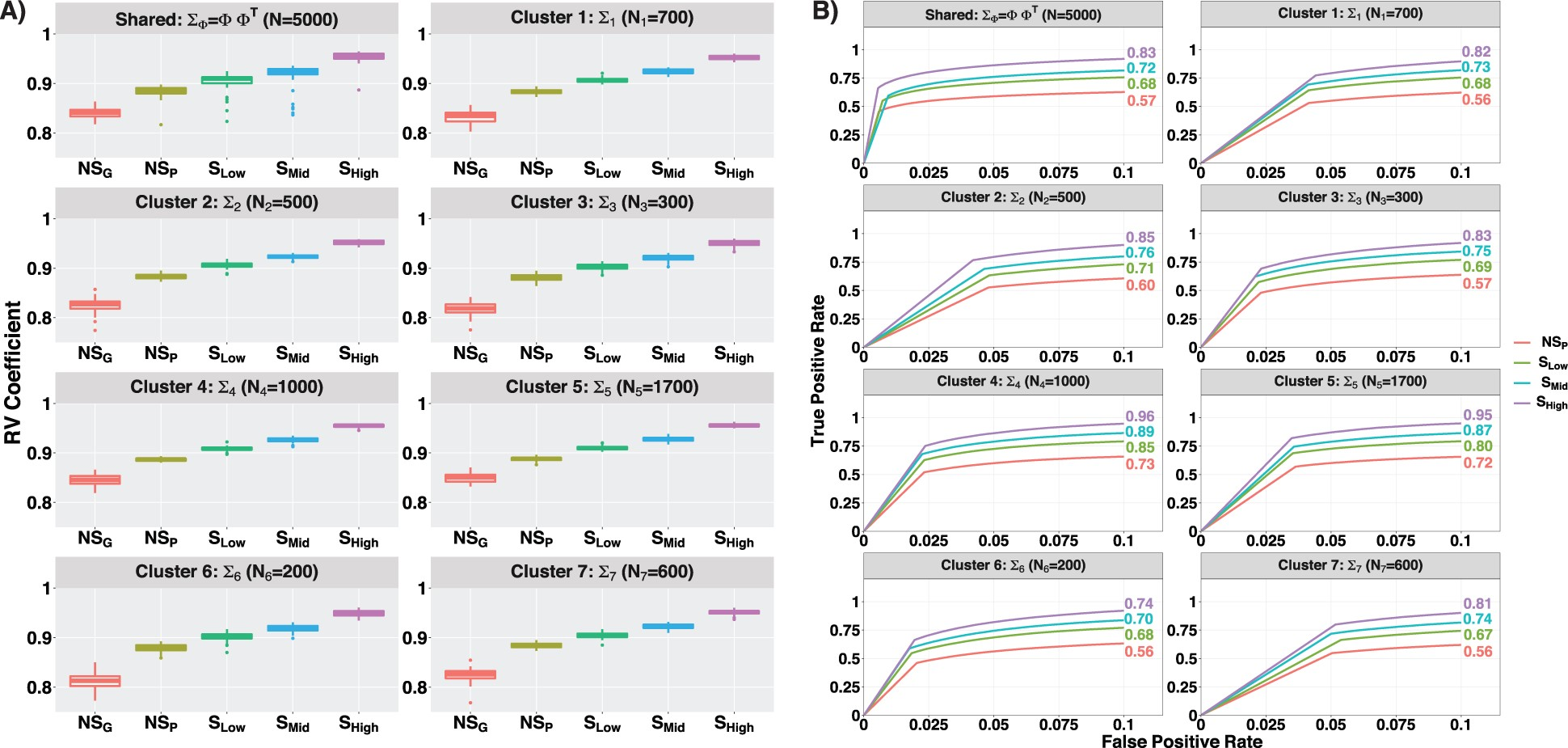
(A)共享的和 cluster 特定的网络表示为
和
。RV 系数衡量不同空间相关程度下真实网络和估计网络之间的距离。在左图中,展示了共享的和特定于 cluster 的网络的 50 个重复的 RV 系数箱线图。基于空间相关性比较了 5 种不同方法的 RV 系数(I)
,(II)
,(III)
,(IV)
,(V)
。(B)在右侧面板中,展示了共享的和特定于 cluster 的网络的 ROC 曲线。Figure 2A 中讨论的五种设置除了
。
网络结构:为了比较网络结构恢复,作者构建了受试者操作特性(ROC)曲线,以比较每个模拟设置检测真实情况的重要边的能力。每个模拟设置的灵敏度(真阳性率)和 1-特异性(假阳性率)在每个阈值参数值 下计算,与 ROC 曲线下面积(AUC)来比较每个模拟设置中的结构恢复(值越高意味着恢复越好)。在 Figure 2B 中,作者提供了前 4 种方法的 ROC 曲线以及 AUCs 关于共享的和 cluster 特定的网络。 方法的 AUC 值的结果低于 0.5,这意味着该方法的表现甚至比随机选择的方法还要差。与网络估计结果类似,从 Figure 2B 中共享的和 cluster 特定的网络的基于 AUC 的比较分析中,观察到较高的空间相关性导致较高的 AUC 值,这意味着更好的网络结构恢复。对于 Figure 2B 中的共享网络,方法 (iv)–(i) 的 AUC 值分别为 0.57、0.68、0.72、0.83,这使得比较方法的准确度提高了 19%、6% 和 15%。
总之,SpaceX 模型显着改善了一系列空间依赖性的网络估计和结构恢复。当空间相关性较高时(例如 0.88),增益最高。这表明,有利地考虑空间相关性以及适当的噪声分布(即泊松模型)可以提高共表达估计的效率。
其他模拟研究:Supplementary Section SB.3 讨论了另一项重复模拟研究,其中生成的数据没有任何空间相关性。将 SpaceX 模型应用于没有任何空间相关性的模拟数据时,没有观察到精度的显着损失。作者进行了另一次重复模拟来研究 hub genes 恢复率,详细信息请参见 Supplementary Section SB.4。作者发现,具有较高连接性的 hub genes 的恢复率高于具有低连接性的 hub genes,并且恢复率随着空间相关性和 cluster 大小的增加而提高。
4 使用空间转录组数据的基因共表达网络
作者使用小鼠下丘脑和人类乳腺癌的两个空间转录组数据集来说明 SpaceX 模型,分别在第 4.1 和 4.2 节中详细介绍。小鼠下丘脑数据集具有单细胞分辨率,spatial locations 代表细胞,location clusters 代表细胞类型。乳腺癌数据集具有区域分辨率,每个空间位置由多个单细胞组成,location clusters 代表三个组织域(肿瘤、中间和正常)。
4.1 下丘脑数据
MERFISH 数据集是从小鼠下丘脑的视前区收集的,该区域调节许多社会行为。MERFISH 技术可测量不同细胞类型的单细胞上的基因表达,从而深入了解组织中细胞的空间组织。该数据集由 160 个基因组成,并在 4812 个 spatial locations 测量相应的基因表达。这些细胞已被注释为 7 种不同的细胞类型(大小),即星形胶质细胞(724)、内皮细胞(503)、室管膜细胞(314)、兴奋性神经元(1024)、抑制性神经元(1694)、未成熟神经元(168) 和成熟神经元神经元(385)。
所有细胞类型的空间分布如 Figure 3A 所示。作者使用 SpaceX 获得共享的和细胞类型特定的网络(Fig. 3B)。共享网络显示在中心,其中基因根据特定细胞类型的差异表达进行分组和颜色编码。作者使用 Wilcoxon 检验来检测某个基因是否针对特定细胞类型进行差异表达。按照 Figure 3B 的所有网络,作者观察到细胞类型内而不是细胞类型之间有更多的 gene-gene 共表达边,这符合预期。为了总结连接水平,作者提供了一个矩阵的圆形热图(Fig. 3C),其中每个条目都是特定细胞类型的基因的基因连接数。右侧细胞类型的树状图显示,未成熟细胞类型中基因之间的连接与其他细胞类型不同。根据每个基因的连接数量,作者确定了每种细胞类型的 hub genes。Figure 3D 通过 upset 图显示了每种细胞类型的 hub genes 和空间交叉点,这是可视化多个集合交叉点的简洁方法。多层维恩图表示前 5 个 hub genes,内部数字表示由 hub genes 控制的基因基数。Supplementary Section SC 中提供了 hub genes 和 top edges 的详细列表。与 MERFISH 数据社区检测(Supplementary Fig. SC.2)和探索性分析(Supplementary Fig. SC.1)相关的发现在 Supplementary Section SC 中讨论。

(A)所有细胞类型和所有主要细胞类型分别的空间分布。图例中提供了细胞类型颜色以及细胞类型信息。每种细胞类型中的 spots 以彩色点显示,而其余 spots 以灰色点显示。(B)共享的和细胞类型特定的网络从 SpaceX 模型中获得。中间的图显示了共享网络,其中 marker genes 根据不同细胞类型的差异表达进行颜色编码。围绕共享网络图提供特定于细胞类型的网络。(C)每个细胞类型特定网络中每个基因连接的圆形热图。基因和细胞类型的树状图分别提供在图的内部和右侧。颜色代表基因连接水平(红色,高;蓝色,中;白色低)。(D)使用 upset 图和多层维恩图分析 hub gene 检测。前 5 个 hub genes 的细胞类型特异性多层维恩图。维恩图中的数字显示了由 hub genes 单独或联合控制的基因基数。upset 图显示了每种细胞类型的不同 hub genes 和不同的空间交叉点。
从 Figure 3D 中,作者发现跨膜蛋白 108(Tmem108)是除内皮细胞之外的所有细胞类型的 hub gene。Tmem 108 蛋白是双相情感障碍和重度抑郁症等精神疾病的主要基因。另外两个检测到的 hub genes CCKBAR 和 CCKBR 作为胆囊收缩素(CCK)的受体,这些基因与胃肠道疾病相关。CCK 受体缺失可导致皮质发育和皮质中间神经元迁移异常。在健康和受伤的小鼠大脑中,sema4D(内皮细胞的另一个 hub gene,未成熟和兴奋性)缺陷会导致少突胶质细胞数量增加。TAC1 响应 ghrelin 管理和性腺功能变化调节肥胖水平。沿着这条线,另一个 hub gene SLN 或肌磷脂的过度表达是肌肉能量的调节剂并减少疲劳。TAC1 和 SLN 在共享的和细胞类型特定的网络中高度相关。这种关联在所有细胞类型中都是保守的,并且这两个基因都是调节肥胖和疲劳的重要因素。
4.2 乳腺癌数据
人类乳腺癌数据是通过对厚度为 16μm 的组织进行活检来收集的。苏木精和伊红(H&E)染色图像如 Figure 4A 左侧所示,其中深色染色代表潜在的肿瘤区域,其余部分可分为中间区域和正常区域。作者根据 H&E 染色图像手动将位置分成三个空间连续的 clusters,包括肿瘤、中间和正常 clusters,clusters 大小分别为 114、67 和 69 个 spots。作者在 Figure 4A 中提供了连续 clusters 的空间分布。表达水平是从 250 个 spot locations 的 5262 个基因测量的,作者使用 SPARK 方法,P-values 的 FDR 截止值为 5%,以检测用于此分析的 290 个空间表达基因。

(A)H&E 染色图像在左侧,手动分类的连续 clusters 的空间分布在右侧。H&E 图像经许可改编自 Ståhl 等人。(B)共享、肿瘤、中间、正常的网络结构,按顺时针方向排列。不同的配色方案已用于表示特定 cluster 的差异表达基因。基因之间的正关联和负关联用不同的线型或颜色表示,而基因之间的关联水平与边宽度成正比。(C)矩阵的圆形热图,每个条目代表特定空间区域的基因连接数。颜色代表基因连接水平(红色,高;蓝色,中;白色低)。基因和空间区域的树状图在热图的内部和右侧给出。(D)多层维恩图显示每个空间区域的前 5 个 hub genes。upset 图列出了每个空间区域和交叉点的所有 hub genes。
作者应用 SpaceX 方法来检测 Figure 4B 中的共享的和 cluster 特定的共表达网络。在共享网络中,如果某个基因针对特定 cluster 有差异表达,作者会使用不同的颜色方案,并为特定于 cluster 的网络保留相同的颜色。作者观察到共享网络比 cluster 特定网络密集得多。根据定义,如果共享结构中的两个基因在特定于 cluster 的网络中关联,则它们之间将存在一定程度的关联,但反之则不然。Figure 4C 显示了每个 cluster 中每个基因的度(连接节点数),cluster 之间的树状图(右侧)显示正常 cluster 中的基因共表达与肿瘤和中间 clusters 中的基因共表达不同,即沿着预期的路线。相应的 circos 图中提供了基因特异性的层次聚类。接下来,作者检测每个 cluster 的 hub genes,并确定所有 clusters 的 hub genes 之间是否存在共性。前 5 个 hub genes 的 cluster 特异性多层维恩图显示了其他基因之间的依赖性。Figure 4D 中相应的 upset 图检测了 cluster 中的共同 hub genes。Supplementary Section SC 提供了用于乳腺癌数据分析的 hub genes 和 top edges 的详细列表。Supplementary Figure SC.4 显示了检测到的共享和细胞类型特异性共表达网络的基因模块。SpaceX 在乳腺癌数据中检测到的许多 hub genes (=19),包括下一段列出的多个胶原蛋白基因,也是另一种基于网络的算法检测到的 hub genes,该算法名为 PRECISE 来自癌症基因组图谱 (TCGA) 的大量乳腺癌数据,支持 SpaceX 的发现(详细信息参见 Supplementary Section SC.5)。
根据作者的分析,多种胶原蛋白基因被检测为肿瘤 clusters 中的 hub genes,例如 COL6A2、COL3A1,它们控制涉及转移的肿瘤迁移。与癌症相关的转录因子、信号通路和受体都可以通过胶原蛋白生物合成来调节。另一个 hub gene,CD24 是一种免疫相关基因,通常在人类肿瘤中过度表达,并调节细胞迁移。VIM 基因(Fig. 4D 中肿瘤和中间区域交叉点之间的 hub gene)可用作癌症早期检测的生物标志物,因为该基因在正常区域中转录不活跃。请注意,SpaceX 不会将 VIM 检测为正常情况的 hub gene。在 Figure 4B 中,作者提供了基因之间的共享网络,其中基因根据每个区域的差异表达用不同的颜色标记。基因 XBP1 是正常生物标志物基因,与肿瘤区域生物标志物基因呈负相关。对于肿瘤网络,作者观察到 LUM 基因与胶原蛋白基因相关,因为 LUM 基因有效调节乳腺癌细胞的雌激素受体和相关功能特性。
5 讨论
作者提出了一种新颖的网络建模方法 SpaceX,它允许根据不同细胞类型或区域的空间转录组数据联合估计共享的和 cluster 特定的网络,从而能够描绘细胞类型或区域的共表达网络的空间异质性。作者通过模拟展示了通过考虑(增加)空间相关性和适当的噪声分布,共表达网络估计和结构的准确性增益。SpaceX 使用小鼠下丘脑和人类乳腺癌数据集中的两个案例研究,可以检测不同细胞类型和肿瘤区域中保守或独特的 top 共表达基因和 hub genes,这些基因具有重要的生物学相关性。特别是,对于小鼠下丘脑数据,作者确定了两个高共表达基因:TAC1 和 SLN,它们与调节体力消耗和体重直接相关。同样,作者确定了多个胶原蛋白基因和 LUM 基因作为乳腺癌数据集的 hub genes,这些基因与癌细胞的关键功能特性(例如肿瘤迁移)相关。这些hub genes 是根据连接到特定基因的边数量来检测的,因此考虑到正在测试大量边,因此自然地考虑了多重性。基于不同中心性的度量[degree centrality by Maslov and Sneppen (2002), betweenness by Freeman et al. (1991) and modularity by Brandes et al. (2007)]可以很容易地适应 SpaceX 方法的网络输出来检测 hub genes。
除了上述两个真实的数据例子外,作者还分析了 Chen 等人关于阿尔茨海默病(AD)的另一个数据,它代表了少数进行基因共表达网络分析的空间转录组学研究之一。AD 数据的原始研究确定了斑块诱导基因 (PIG) 的多细胞基因共表达网络,其中包括补体系统、氧化应激、溶酶体和炎症,这是 AD 后期的特征。作者应用 SpaceX 方法在相同数据中检测 hub PIGs,并能够检测到原始研究中发现的具有高共表达的 top hub genes 的很大一部分,这凸显了 SpaceX 的强大功能。有关此分析的更详细描述可以在 Supplementary Section SC.6 中找到。
SpaceX 核心方法可以概括为几个方向。SpaceX 的模型可以适应其他噪声分布,例如负二项式或其他稳健分布,以推断不同平台的空间共表达网络。此外,可以容纳多个空间内核来建模平稳和非平稳相关结构,以丰富推理。所提出的方法基于监督聚类,未来可以扩展到无监督聚类技术。所提出的方法有可能扩展到研究不同生物系统中的依赖性,例如蛋白质之间的结合或疾病特异性基因共表达。作者提出了模型的修改版本,以进一步解释细胞类型特定 cluster 之间的空间相关性(更多详细信息,请参阅 Supplementary Section SA.6)。
可能驱动空间相关性的一个潜在因素是总 read 深度。每个位置的总 read 深度(在等式(1)中用 M 表示)可能会因组织切片的渗透性水平不同以及随后对 mRNA 的可及性而因位置而异,从而导致某些组织区域的总 read 深度较高而其他区域总 read 深度较低。该技术因素可能导致相关 reads,相关性取决于相邻细胞的细胞属性。在这种情况下,归一化常数 M 或 read 深度可以基于类似的细胞邻域细胞属性与内在速率相关,并且如果有相关 read 计数的证据,也可以使用空间过程对 M 进行建模。
SpaceX 采用高效的降维技术,在具有单 CPU 核心的高计算集群中运行乳腺癌和小鼠下丘脑数据集大约需要 1.5 小时和 5 小时。目前,作者的方法仅限于数百个基因,随着技术的成熟,作者的目标是将方法扩展到数千个基因和 spots 的数量。SpaceX 包和补充材料可分别在 github.com/bayesrx/SpaceX 和 bookdown.org/satwik91/SpaceX_supplementary/ 上获取。
注:本文为个人学习笔记,仅供大家参考学习,不得用于任何商业目的。如有侵权,请联系作者删除。

本文由 mdnice 多平台发布















 698
698











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









