









1、引言
meconetcomp包是microeco包的扩展包。两个包均由福建农林大学姚敏杰教授团队开发,专注于扩增子测序数据的集成分析。Meconetcomp发表在国人创建的顶级生物信息学期刊iMeta中。其中meconetcomp包提供了一种比较微生物共现网络的流程,具有较高的灵活性和可扩展性,可以帮助用户高效地比较不同组样本或不同构建方法构建的网络。(一行代码可实现某个特性分析),因此,掌握了这套代码,可让你快速通关网络分析。

1.1、meconetcomp包和microeco分析流程比较
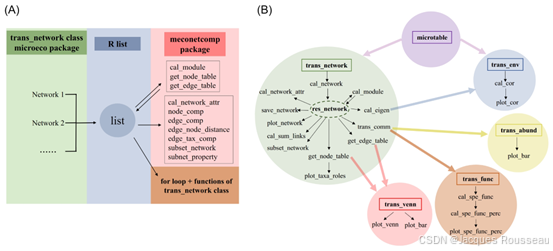
图片源自姚敏杰教授imate文章。图1、本方案所用方法的示意图。(A)整个方案的示意图。(B)由方案中微生态包的相关类和功能组成的流程图。方框表示类。箭头表示数据分析中的定向流。没有方框的文本表示相应类中的功能
1.2、代码输出结果欣赏
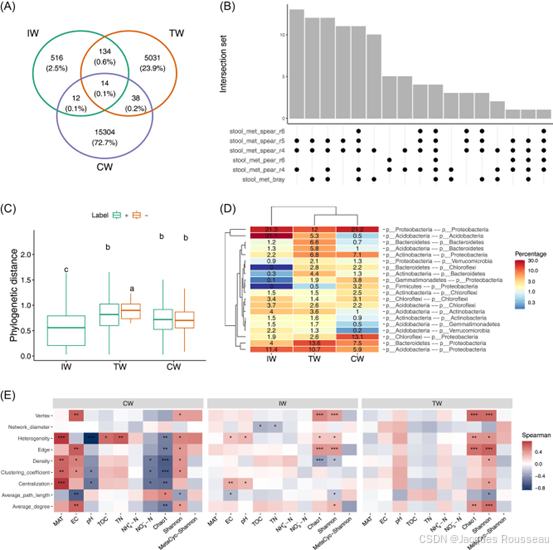
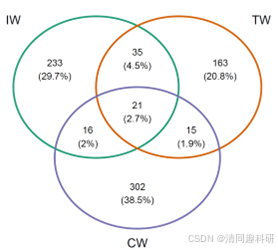
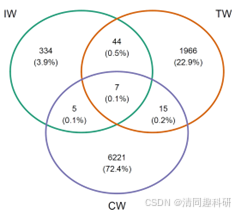
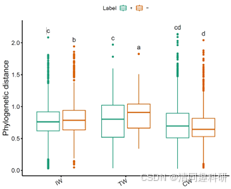
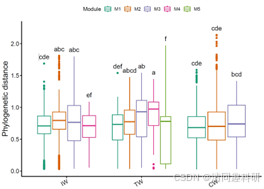
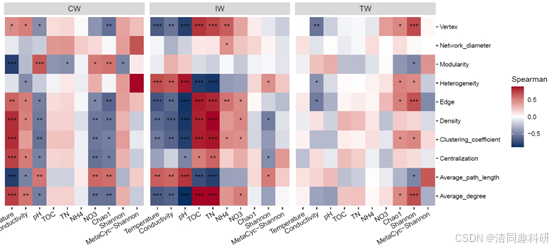
图2、流程中部分分析结果的可视化:(A)soil_amp_network 中三个网络之间边的维恩图。(B)soil_met_network 中具有不同构建方法的所有网络的边的交集。(C)soil_amp_network 中边节点之间的系统发育距离。标签“+”代表正边,即具有正关联的边。标签“-”表示负边,即具有负关联的边。不同的小写字母表示在使用 agricolae 包的 duncan.test 函数进行 Duncan 新多重极差检验的方差分析中不同组之间的显著差异(p < 0.05)。(D)soil_amp_network 网络中正边链接节点所属门的数量分布。图中的数字表示边与网络中的所有正边的比例。(E)子网络属性与非生物因素/多样性之间的相关性。 * p < 0.05, ** p < 0.01; *** p < 0.001。CW,滨海湿地;EC,电子电导率;IW,中国内陆湿地;MAT,年平均气温;MetaCyc-Shannon,基于湿地土壤宏基因组测序的MetaCyc通路丰度的Shannon熵;TN,总氮;TOC,总有机碳;TW,青藏高原湿地。
2、示例数据和R代码
2.1、数据准备
准备数据
⚠️该论文中读取数据仅一句简单的data(soil_amp)就直接加载了所需要分析的数据,所以对于有需求的朋友,想用自己的数据就显得十分困难。
经过小编对soil_amp.R数据进行解析发现该R数据集是采用microeco包中建立微表的方式构建的,我们解析后发现了该微表共输入了四个数据分别是otu表(feature_table.csv),样本信息表(sample_table.csv),物种分类表(tax_table.csv)和系统发育树文件(phylo_tree.tre)。
⚠️一般数据输入采用上述4个表格会流畅的获得相关结果,但是由于数据来源的差异,部分数据表格可能没有,因此经测试最少需要otu表(feature_table.csv),样本信息表(sample_table.csv)可满足大部分输出结果。
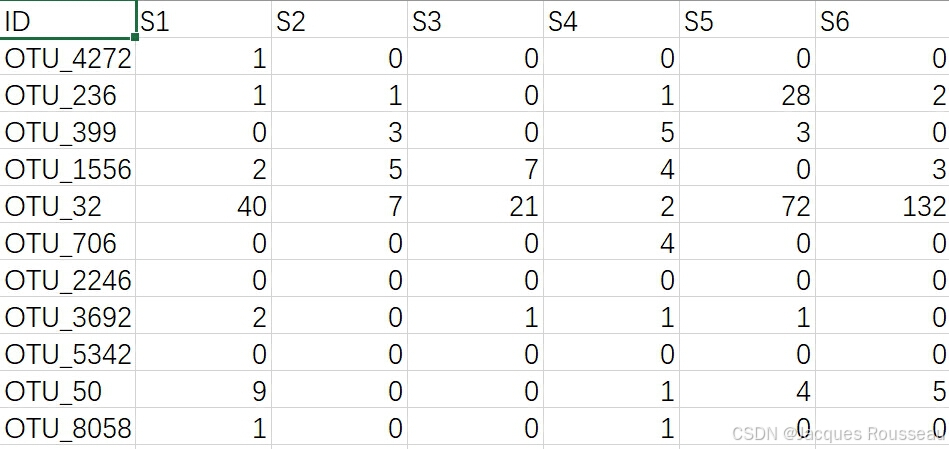
(1)otu表(feature_table.csv)
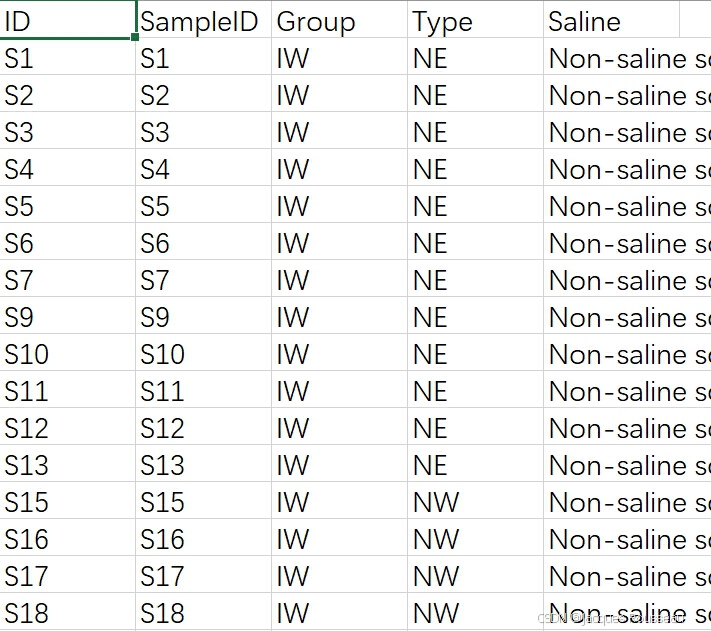
(2)样本信息表(sample_table.csv)
(3)物种分类表(tax_table.csv)
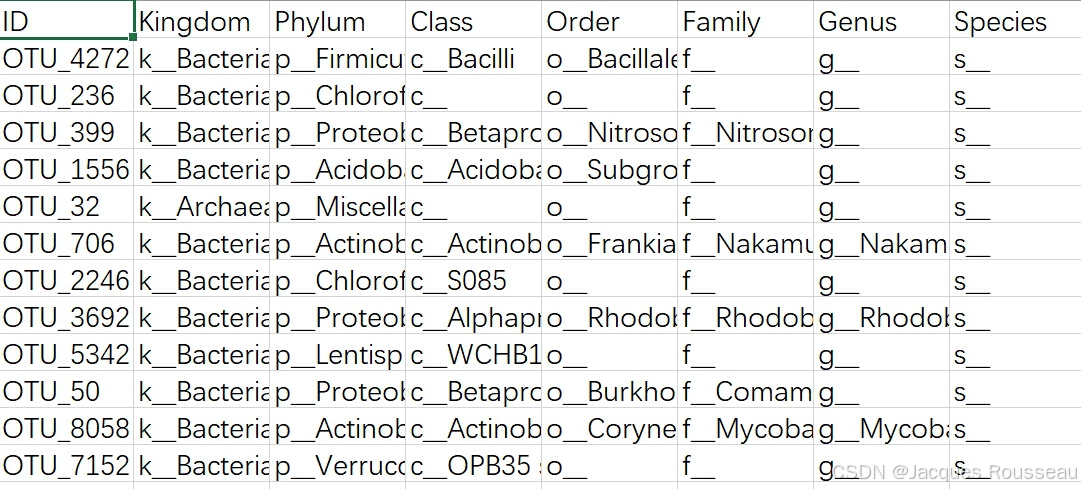
(4)系统发育树文件(phylo_tree.tre)
2.2、R包准备和数据读取
🌟基础操作之meconetcomp包等依赖包安装
# 安装所需的 R 包
# aplot: microeco 包中 trans_venn 类的依赖包
# agricolae: 用于 Duncan 的新多重范围检验
packages <- c("meconetcomp", "rgexf", "pheatmap", "aplot", "agricolae")
# 检查每个包是否已安装,如果未安装则安装
for(x in packages) {
if(!require(x, character.only = TRUE)) {
install.packages(x, dependencies = TRUE) # 自动安装包及其所有依赖项
}
}
###########数据读取和构建微表#############
#安装并加载ape包
if(!require("ape")){install.packages("ape")}
library(ape)
##################数据基本操作#######################
# 加载示例数据;16S rRNA基因扩增子测序数据
#读取样本分组信息表
sample_info_16S <- read.csv("sample_table.csv", row.names = 1)
#读取特征表
otu_table_16S <- read.csv("feature_table.csv",row.names = 1)
#读取系统发育树
phylo_tree_16S <- read.tree("phylo_tree.tre", tree.names = NULL)
#读取物种分类表
taxonomy_table_16S <- read.csv("tax_table.csv",row.names = 1)
# 让我们创建需要信息的 microtable 对象
soil_amp <- microtable$new(sample_table = sample_info_16S, otu_table = otu_table_16S, tax_table = taxonomy_table_16S, phylo_tree = phylo_tree_16S)
2.3、创建共现性网络
⚠️本示例提供了3个组别,可根据自己的数据进行设计。
# 选择"IW"组的样本
# 使用clone对soil_amp对象深度拷贝
tmp <- clone(soil_amp)
# 直接更改样本表
tmp$sample_table %<>% subset(Group == "IW")
# 清理对象中的所有文件
tmp$tidy_dataset()
# 使用filter_thres参数过滤低丰度特征
tmp <- trans_network$new(dataset = tmp, cor_method = "spearman", filter_thres = 0.0005)
# 设置 p 值阈值
# 设置相关系数阈值
tmp$cal_network(COR_p_thres = 0.01, COR_cut = 0.6)
# 将网络加入列表中
soil_amp_network$IW <- tmp
# 选择"TW"组的样本
tmp <- clone(soil_amp)
tmp$sample_table %<>% subset(Group == "TW")
tmp$tidy_dataset()
tmp <- trans_network$new(dataset = tmp, cor_method = "spearman", filter_thres = 0.0005)
tmp$cal_network(COR_p_thres = 0.01, COR_cut = 0.6)
soil_amp_network$TW <- tmp
# 选择"CW"组的样本
tmp <- clone(soil_amp)
tmp$sample_table %<>% subset(Group == "CW")
tmp$tidy_dataset()
tmp <- trans_network$new(dataset = tmp, cor_method = "spearman", filter_thres = 0.0005)
tmp$cal_network(COR_p_thres = 0.01, COR_cut = 0.6)
soil_amp_network$CW <- tmp
# 现在已创建网络列表 soil_amp_network
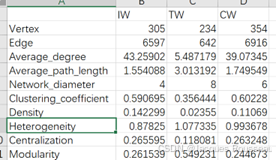
2.4、网络拓扑属性计算
###########网络模块化计算######
soil_amp_network %<>% cal_module(undirected_method = "cluster_fast_greedy")
###########网络拓扑属性计算###########
tmp <- cal_network_attr(soil_amp_network)
# tmp 是一个包含网络属性的数据框,输出tmp文件
write.csv(tmp,"tmp.csv")
##############提取所有网络的节点和边属性##########
soil_amp_network %<>% get_node_table(node_roles = TRUE) %>% get_edge_table
输出结果:
2.5、多网络特性比较
🌟跨网络比较节点
比较网络重叠节点和差异节点
# 使用 meconetcomp 包中的 node_comp 函数将所有网络中的节点转换为一个新表对象,然后使用 trans_venn 类轻松分析节点重叠情况。
tmp <- node_comp(soil_amp_network, property = "name") # 获取节点分布
tmp1 <- trans_venn$new(tmp, ratio = "numratio") # 获取节点交集
g1 <- tmp1$plot_venn(fill_color = FALSE) # 绘制韦恩图
g1
ggsave("soil_amp_node_overlap.pdf", g1, width = 7, height = 6)
# 计算 Jaccard 距离以反映网络的整体差异
tmp$cal_betadiv(method = "jaccard")
tmp$beta_diversity$jaccard
🌟跨网络比较边
# 获取跨网络的边分布
tmp <- edge_comp(soil_amp_network)
tmp1 <- trans_venn$new(tmp, ratio = "numratio") # 获取边的交集
g1 <- tmp1$plot_venn(fill_color = FALSE)
ggsave("soil_amp_edge_overlap.pdf", g1, width = 7, height = 6)
# 计算 Jaccard 距离
tmp$cal_betadiv(method = "jaccard")
tmp$beta_diversity$jaccard
🌟将网络重叠的边提取到新网络
# 获取边的分布和交集
tmp <- edge_comp(soil_amp_network)
tmp1 <- trans_venn$new(tmp)
# 将交集结果转换为 microtable 对象
tmp2 <- tmp1$trans_comm()
# 提取所有三组网络 ("IW", "TW" 和 "CW") 的交集
Intersec_all <- subset_network(soil_amp_network, venn = tmp2, name = "IW&TW")
Intersec_all$save_network("Intersec_all.gexf") # 保存为 gexf 格式
🌟比较边中成对节点的系统发育距离
# 过滤低丰度特征以加速计算
node_names <- unique(unlist(lapply(soil_amp_network, function(x) { colnames(x$data_abund) })))
filter_soil_amp <- microeco::clone(soil_amp)
filter_soil_amp$otu_table <- filter_soil_amp$otu_table[node_names, ]
filter_soil_amp$tidy_dataset()
# 获取系统发育距离矩阵
phylogenetic_distance_soil <- as.matrix(cophenetic(filter_soil_amp$phylo_tree))
# 使用正负标签进行计算
tmp <- edge_node_distance$new(network_list = soil_amp_network, dis_matrix = phylogenetic_distance_soil, label = c("+", "-"))
tmp$cal_diff(method = "anova")
# 可视化
g1 <- tmp$plot(add = "none", add_sig = TRUE, add_sig_text_size = 5) + ylab("Phylogenetic distance")
ggsave("soil_amp_phylo_distance.pdf", g1, width = 7, height = 6)
# 显示具有至少 10 个节点和正边的不同模块
tmp <- edge_node_distance$new(network_list = soil_amp_network, dis_matrix = phylogenetic_distance_soil,
label = "+", with_module = TRUE, module_thres = 10)
tmp$cal_diff(method = "anova")
g1 <- tmp$plot(add = "none", add_sig = TRUE, add_sig_text_size = 5) + ylab("Phylogenetic distance")
g1
ggsave("soil_amp_phylo_distance_modules.pdf", g1, width = 8, height = 6)
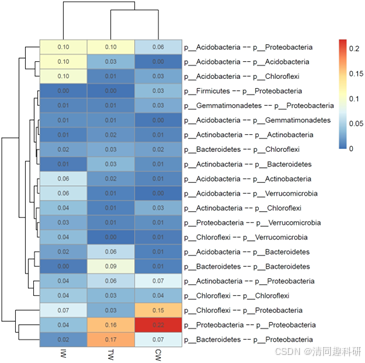
🌟比较跨网络边的节点来源
soil_amp_network_edgetax <- edge_tax_comp(soil_amp_network, taxrank = "Phylum", label = "+", rel = TRUE)
# 过滤小数量特征
soil_amp_network_edgetax <- soil_amp_network_edgetax[apply(soil_amp_network_edgetax, 1, mean) > 0.01, ]
# 可视化
g1 <- pheatmap::pheatmap(soil_amp_network_edgetax, display_numbers = TRUE)
g1
ggsave("soil_amp_edge_tax_comp.pdf", g1, width = 7, height = 7)
🌟比较子网络的拓扑性质
#该部分首先根据soil_amp数据集中每个样本所含的OTU提取soil_amp_network中每个网络的子网络,然后计算子网络的全局拓扑性质,所有操作都封装在meconetcomp包的subnet_property函数中。
# 计算所有子网络的全局属性
tmp <- subnet_property(soil_amp_network)
# 为相关性分析准备数据
# 将第二列(样本名)作为行名
rownames(tmp) <- tmp[, 2]
# 删除前两列(网络名称和样本名称)
tmp <- tmp[, -c(1:2)]
# 加载现成的环境因子和多样性数据表
data(soil_measure_diversity)
# 创建 trans_env 对象,添加环境因子和多样性数据
tmp1 <- trans_env$new(dataset = soil_amp, add_data = soil_measure_diversity)
# 计算相关性矩阵,按组分析,使用添加的丰度表 tmp,Spearman 方法
tmp1$cal_cor(use_data = "other", by_group = "Group", add_abund_table = tmp, cor_method = "spearman")
# 生成相关性热图
g1 <- tmp1$plot_cor()
# 保存热图至 PDF 文件
ggsave("soil_amp_subnet_property.pdf", g1, width = 11, height = 5)
输出结果:
3、参考文献
[1] Liu, Chi, Chaonan Li, Yanqiong Jiang, Raymond J. Zeng, Minjie Yao, and Xiangzhen Li. 2023. “ A guide for comparing microbial co-occurrence networks.” iMeta 2, e71.
[2] Chi Liu, Yaoming Cui, Xiangzhen Li, Minjie Yao, microeco: an R package for data mining in microbial community ecology, FEMS Microbiology Ecology, Volume 97, Issue 2, February 2021, fiaa255
[3] microeco_tutorial/meconetcomp-package.html#network-modularity-for-all-networks
4、联系作者
!!!有需要的小伙伴可以去评论区自取今天的测试代码和实例数据。
📌示例代码中提供了数据和代码,已经小编测试,可直接运行。
⚠️本账号会不时更新代码,代码失效可联系小编~
以上就是利用R语言microeco包和meconetcomp包解析网络特性和多网络比较的全部内容。

















 531
531

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









