






目录
1、引言
R包iCAMP为群落组装机制的量化分析提供了一种新的方法。基于系统发育分箱的零模型分析,用于评估不同生态过程(同质选择、异质选择、扩散和漂变)对整个群落及各个系统发育分箱(bin)的相对重要性。Ning et al. 2020年在Nature Communications发布该方法以来截止2024年9月已经被引506次,成为了群落构建分析的重要分析方法。该方法也成为nature,science系列文章的常客,深受大家喜欢,在文章中运用该方法将为论文增色不少。
2、文献美图欣赏
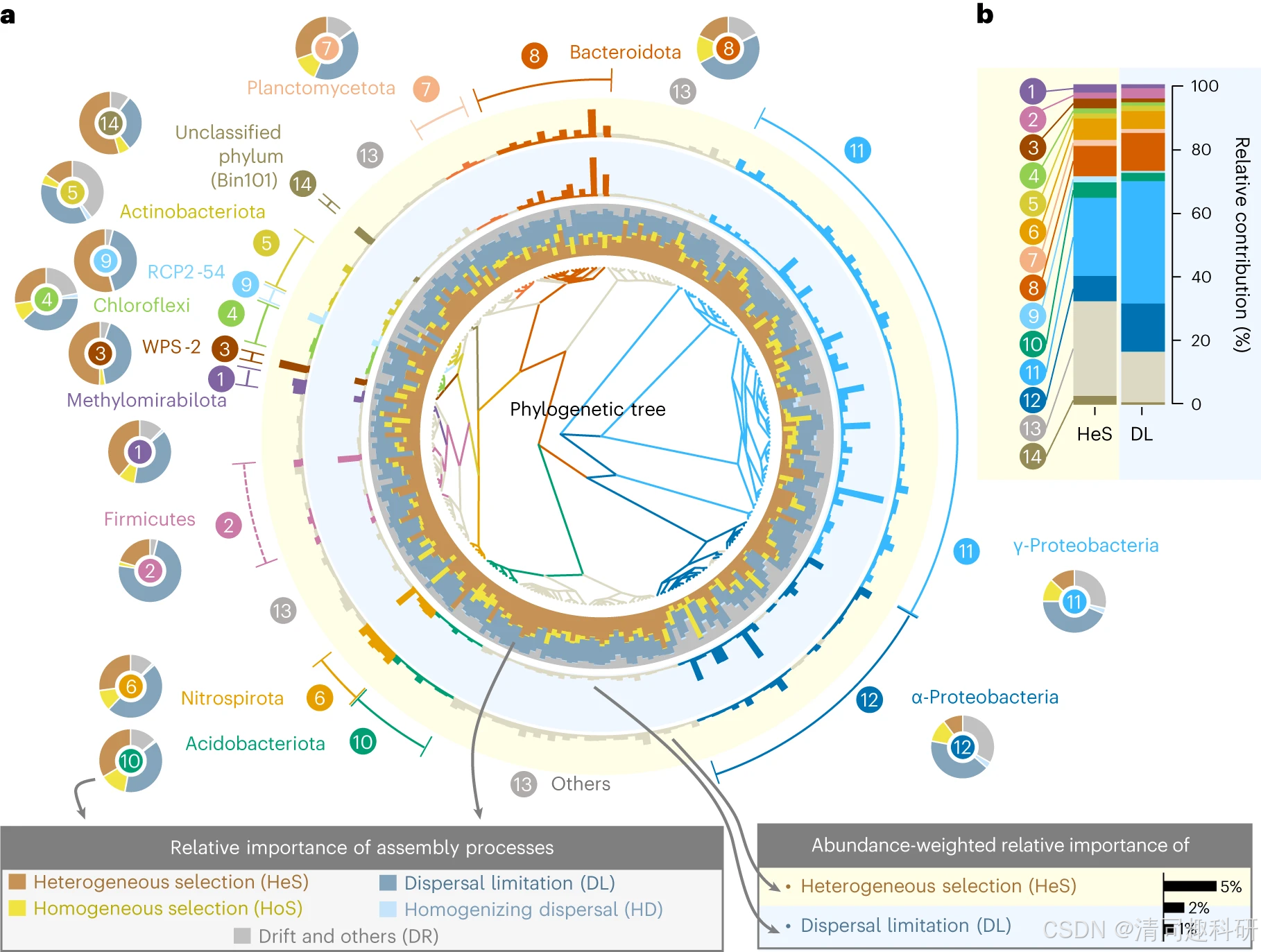
图片来源,Ning et al. 2020,Nature Communications。
中央是系统发育树,内环展示了不同过程的相对重要性,表示每个系统发育箱中的DL(中间环)和HeS(外环)的丰度加权重要性。外环和内环的圆环图显示了每个门(或变形菌门的分类)中不同过程的相对重要性。
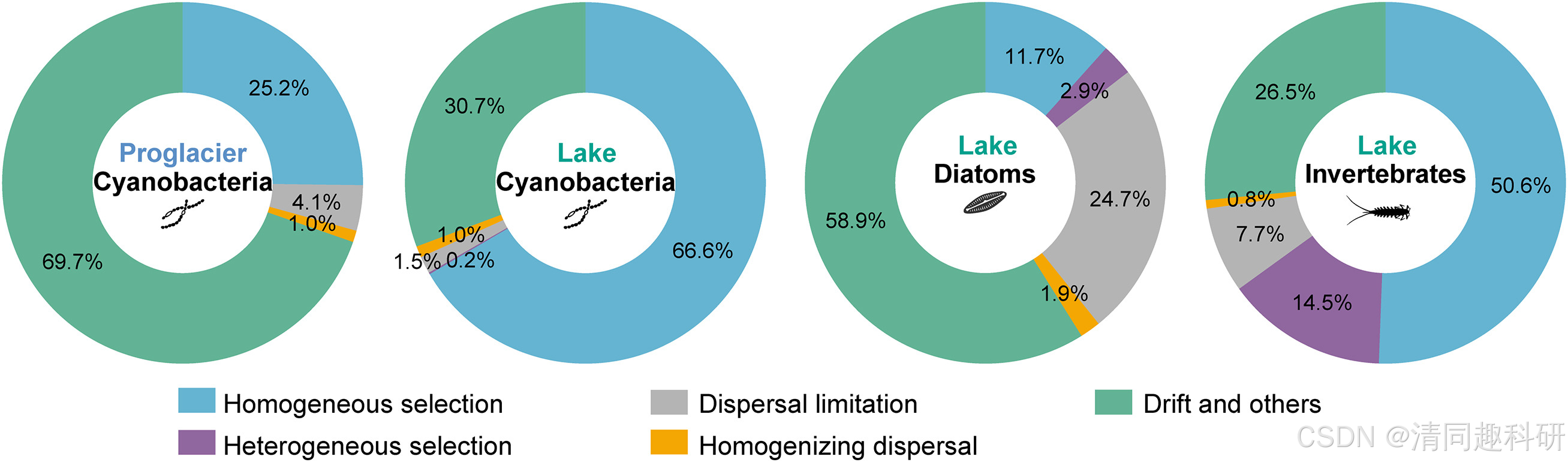
图片来源,Ning et al. 2020,Nature Communications。
不同群落组装过程在冰川前缘和湖泊栖息地中对蓝藻、硅藻和无脊椎动物的相对重要性。
3、iCAMP分析流程
iCAMP的核心功能是通过系统发育分箱的零模型分析,量化不同生态过程的相对重要性。将分类群依据其系统发育关系划分到不同的bin。核心参数是βNRI和RC。βNRI值小于-1.96或大于1.96分别指示同质选择(HoS)和异质选择(HeS);当|βNRI|≤1.96且RC值小于-0.95时,表示均质扩散(HD);当|βNRI|≤1.96且RC值大于0.95时,表示扩散限制(DL);当|βNRI|≤1.96且|RC|≤0.95时,表示漂变(DR)。
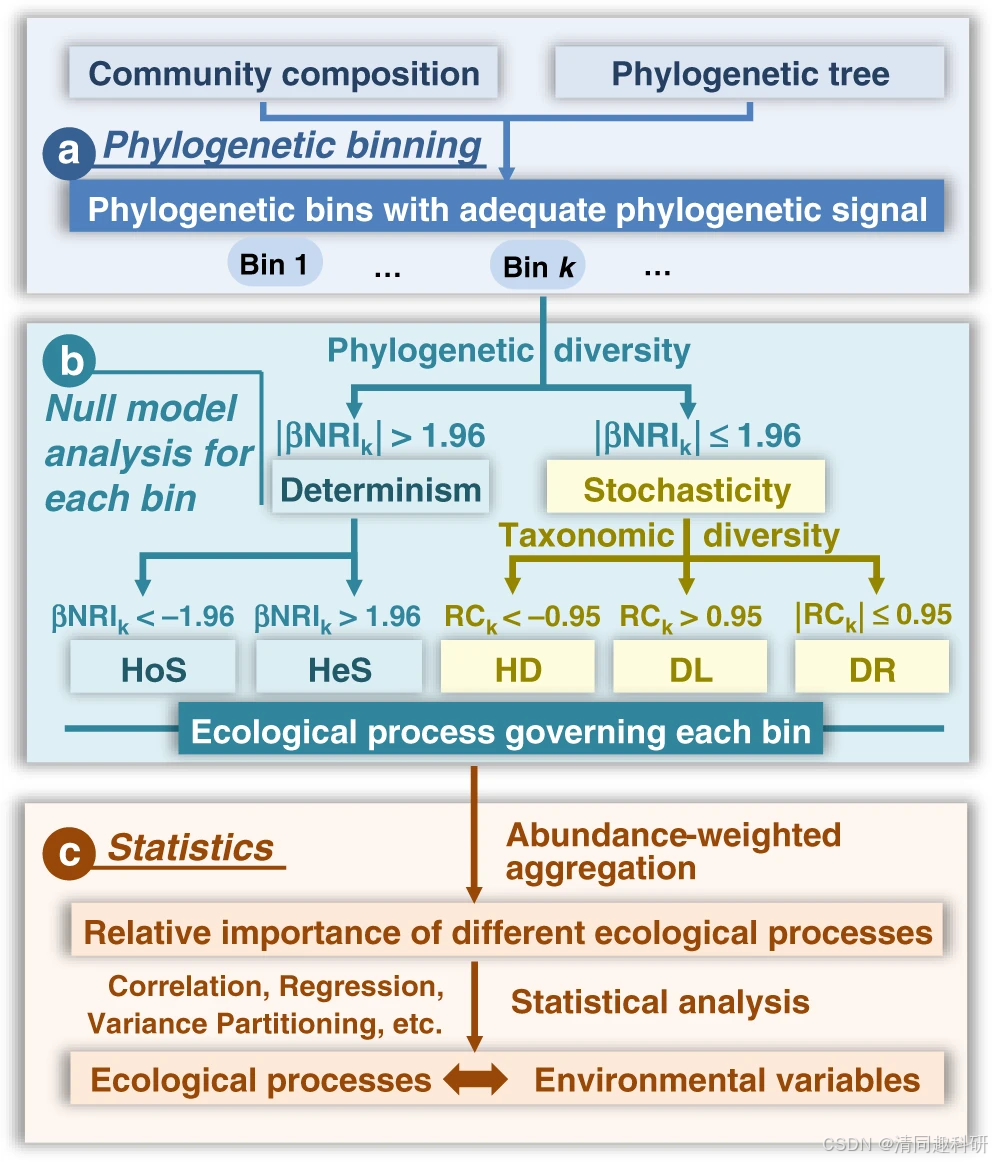
图片来源,Ning et al. 2020,Nature Communications。
4、示例数据和R代码
4.1、数据准备
下述文件1,2,4为必须文件,3和5可以根据研究目标进行取舍
1、OTU 表文件(制表符分隔的 TXT 文件):otus.txt
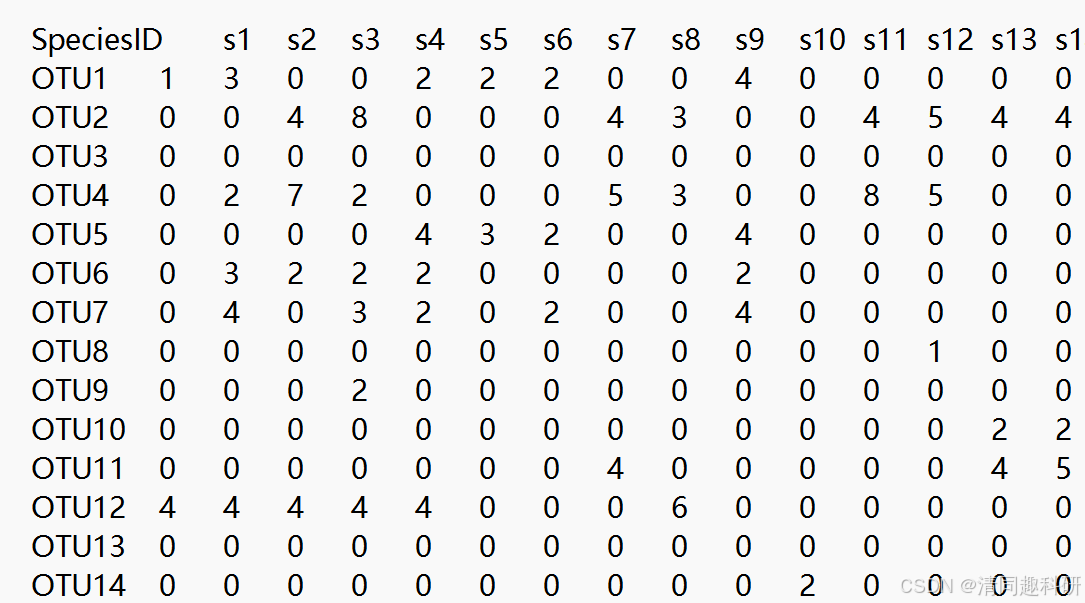
2、系统发育树文件:tree.nwk
3、分类(分类学)信息:classification.txt

4、处理信息表:treat2col.txt

5、环境变量:environment.txt

4.2、完整R代码
设置文件夹路径和文件名
# 1 # 设置文件夹路径和文件名,请根据计算机上的实际文件夹路径和文件名进行更改。
# 保存输入文件的文件夹
wd="E:/0A博士项目/11参与项目/生信项目/零模型/icamp1"
# OTU 表文件(制表符分隔的 TXT 文件)
com.file <- "otus.txt"
# 系统发育树文件
tree.file <- "tree.nwk"
# 分类(分类学)信息
clas.file <- "classification.txt"
# 处理信息表
treat.file <- "treat2col.txt"
# 环境变量
env.file <- "environment.txt"
# 如果没有环境文件或环境变量不代表生态位,可以跳过第7步和第8步。
# 但要检查确定 binning 设置的替代方法,例如 bin.size.limit。
# 环境变量文件路径可能与实际情况不同,所以需要根据实际情况调整
# 保存输出的文件夹路径。即使只是测试示例数据,也请更改为一个新的文件夹。
save.wd="E:/0A博士项目/11参与项目/生信项目/零模型/icamp1/Testoutput2"
if(!dir.exists(save.wd)){dir.create(save.wd)}
设置参数
# 2 # 关键参数设置
prefix <- "Test" # 输出文件名的前缀。通常使用项目 ID。
rand.time <- 100 # 随机化次数,1000 次通常足够。例如测试时,可以设置为 100 或更少以节省时间。
nworker <- 4 # nworker 是并行计算的线程数,这取决于计算机的 CPU 核心数。
#memory.G <- 50 # 设置所需的内存大小(新版本R无法运行该函数)
载入包并读取数据
# 3 # 载入包并读取数据
if (!require(iCAMP, quietly = TRUE)) install.packages("iCAMP")
if (!require(ape, quietly = TRUE)) install.packages("ape")
library(iCAMP)
library(ape)
setwd(wd)
comm=t(read.table(com.file, header = TRUE, sep = "\t", row.names = 1,
as.is = TRUE, stringsAsFactors = FALSE, comment.char = "",
check.names = FALSE))
tree=read.tree(file = tree.file)
clas=read.table(clas.file, header = TRUE, sep = "\t", row.names = 1,
as.is = TRUE, stringsAsFactors = FALSE, comment.char = "",
check.names = FALSE)
treat=read.table(treat.file, header = TRUE, sep = "\t", row.names = 1,
as.is = TRUE, stringsAsFactors = FALSE, comment.char = "",
check.names = FALSE)
env=read.table(env.file, header = TRUE, sep = "\t", row.names = 1,
as.is = TRUE, stringsAsFactors = FALSE, comment.char = "",
check.names = FALSE) # skip this if you do not have env.file
匹配OUT表和信息表以及树文件中样本ID
# 4 # 匹配 OTU 表和处理信息表中的样本 ID
sampid.check = match.name(rn.list = list(comm = comm, treat = treat, env = env))
# sampid.check = match.name(rn.list = list(comm = comm, treat = treat)) # 如果没有 env 文件
# 对于示例数据,输出应为 "All match very well"。
# 对于您的数据文件,如果未匹配 ID,则未匹配的样本将被删除。
treat = sampid.check$treat
comm = sampid.check$comm
comm = comm[, colSums(comm) > 0, drop = FALSE] # 如果某些不匹配的样本被移除,可能会出现一些虚假的 OTU,您可以使用这一行代码在必要时将它们移除。
env = sampid.check$env # 如果没有 env 文件则跳过此步骤
# 5 # 匹配 OTU 表和树文件中的 OTU ID
spid.check = match.name(cn.list = list(comm = comm), rn.list = list(clas = clas), tree.list = list(tree = tree))
# 对于示例数据,输出应为 "All match very well"。
# 对于您的数据文件,如果之前没有匹配 ID,则未匹配的 OTU 将被删除。
comm = spid.check$comm
clas = spid.check$clas
tree = spid.check$tree
计算成对的系统发育距离矩阵
# 由于微生物群落数据通常包含大量物种(OTU 或 ASV),我们使用 R 包 "bigmemory" 中的 "big.matrix" 来处理大型系统发育距离矩阵。
setwd(save.wd)
if (!file.exists("pd.desc"))
{
pd.big = iCAMP::pdist.big(tree = tree, wd = save.wd, nworker = nworker)
# 输出文件:
# path.rda: 一个 R 对象,列出从根到每个叶子的所有节点和边长,保存为 R 数据格式。这是计算系统发育距离矩阵的一个中间输出。
# pd.bin: BIN 文件(backingfile),由 R 包 bigmemory 中的 big.matrix 函数生成。这是存储成对系统发育距离值的大矩阵。使用这种 bigmemory 格式文件时,计算时不需要物理内存,而是使用硬盘。
# pd.desc: DESC 文件(描述文件),用于保存 backingfile(pd.bin)的描述。
# pd.taxon.name.csv: 逗号分隔的 CSV 文件,存储树叶的 ID(OTU),用作大系统发育距离矩阵的行/列名称。
} else {
# 如果您已经在之前的运行中计算了系统发育距离矩阵
pd.big = list()
pd.big$tip.label = read.csv(paste0(save.wd, "/pd.taxon.name.csv"), row.names = 1, stringsAsFactors = FALSE)[, 1]
pd.big$pd.wd = save.wd
pd.big$pd.file = "pd.desc"
pd.big$pd.name.file = "pd.taxon.name.csv"
}
确定bin内最小物种数目
# 7 # 评估物种之间的生态位偏好差异
# 这个步骤需要 env 文件。
# 由于微生物群落数据通常包含大量物种(OTU 或 ASV),我们使用 R 包 "bigmemory" 中的 "big.matrix" 来处理大型生态位差异矩阵。
setwd(save.wd)
niche.dif = iCAMP::dniche(env = env, comm = comm, method = "niche.value",
nworker = nworker, out.dist = FALSE, bigmemo = TRUE,
nd.wd = save.wd)
# 8 # 评估每个分类单元内的系统发育信号
# 对于实际数据,你可以尝试不同的分箱设置,并选择能够产生最佳分类单元内系统发育信号的设置。
# 这个步骤需要 env 文件。
# 8.1 # 尝试使用当前设置进行系统发育分箱
ds = 0.2 # 设置可以更改以探索最佳选择
bin.size.limit = 5 # 设置可以更改以探索最佳选择。此处设置为5,仅用于小型示例数据集。对于实际数据,通常尝试12到48。
# 对于 taxa.binphy.big 函数,树必须是有根的。
if(!ape::is.rooted(tree))
{
tree.rt = iCAMP::midpoint.root.big(tree = tree, pd.desc = pd.big$pd.file,
pd.spname = pd.big$tip.label, pd.wd = pd.big$pd.wd,
nworker = nworker)
tree = tree.rt$tree
}
phylobin = taxa.binphy.big(tree = tree, pd.desc = pd.big$pd.file, pd.spname = pd.big$tip.label,
pd.wd = pd.big$pd.wd, ds = ds, bin.size.limit = bin.size.limit,
nworker = nworker)
# 8.2 # 测试分类单元内的系统发育信号
sp.bin = phylobin$sp.bin[, 3, drop = FALSE] # 从 phylobin 中提取系统发育分类单元
sp.ra = colMeans(comm / rowSums(comm)) # 计算每个物种的相对丰度
abcut = 3 # 你可以移除一些物种,如果它们太稀有而无法进行可靠的相关性测试
commc = comm[, colSums(comm) >= abcut, drop = FALSE] # 只保留丰度大于 abcut 的物种
dim(commc) # 输出 commc 的维度
spname.use = colnames(commc) # 获取物种名称
binps = iCAMP::ps.bin(sp.bin = sp.bin, sp.ra = sp.ra, spname.use = spname.use,
pd.desc = pd.big$pd.file, pd.spname = pd.big$tip.label, pd.wd = pd.big$pd.wd,
nd.list = niche.dif$nd, nd.spname = niche.dif$names, ndbig.wd = niche.dif$nd.wd,
cor.method = "pearson", r.cut = 0.1, p.cut = 0.05, min.spn = 5) # 计算系统发育信号
# 将结果保存到文件中
if(file.exists(paste0(prefix, ".PhyloSignalSummary.csv"))) {
appendy = TRUE
col.namesy = FALSE
} else {
appendy = FALSE
col.namesy = TRUE
}
write.table(data.frame(ds = ds, n.min = bin.size.limit, binps$Index),
file = paste0(prefix, ".PhyloSignalSummary.csv"),
append = appendy, quote = FALSE, sep = ",", row.names = FALSE, col.names = col.namesy)
if(file.exists(paste0(prefix, ".PhyloSignalDetail.csv"))) {
appendy2 = TRUE
col.namesy2 = FALSE
} else {
appendy2 = FALSE
col.namesy2 = TRUE
}
write.table(data.frame(ds = ds, n.min = bin.size.limit, binID = rownames(binps$detail), binps$detail),
file = paste0(prefix, ".PhyloSignalDetail.csv"),
append = appendy2, quote = FALSE, sep = ",", row.names = FALSE, col.names = col.namesy2)
# 由于这个示例小数据是随机生成的,相关性应该非常弱。
# 通常,你需要寻找一个能够导致更高的 RAsig.abj(具有显著系统发育信号的分类单元的相对丰度)
#和相对较高的 meanR(分类单元间的平均相关系数)的分箱设置。
# 详细请参阅 "ps.bin" 函数的帮助文档以了解输出的意义。
iCAMP分析
# 9 # iCAMP分析
# 9.1 # 不去除小分类单元
# 通常使用 # 将 sig.index 设置为 Confidence,而不是 SES.RC (betaNRI/NTI + RCbray)
bin.size.limit = 5 # 对于真实数据,通常根据系统发育信号测试选择适当的数字或尝试一些设置,然后选择合理的随机性水平。我们的经验值是 12、24 或 48。但由于这个示例数据集太小,只能使用 5。
sig.index = "Confidence" # 参考 icamp.big 帮助文档中的其他选项。
icres = iCAMP::icamp.big(comm = comm, pd.desc = pd.big$pd.file, pd.spname = pd.big$tip.label,
pd.wd = pd.big$pd.wd, rand = rand.time, tree = tree,
prefix = prefix, ds = 0.2, pd.cut = NA, sp.check = TRUE,
phylo.rand.scale = "within.bin", taxa.rand.scale = "across.all",
phylo.metric = "bMPD", sig.index = sig.index, bin.size.limit = bin.size.limit,
nworker = nworker, rtree.save = FALSE, detail.save = TRUE,
qp.save = FALSE, detail.null = FALSE, ignore.zero = TRUE, output.wd = save.wd,
correct.special = TRUE, unit.sum = rowSums(comm), special.method = "depend",
ses.cut = 1.96, rc.cut = 0.95, conf.cut = 0.975, omit.option = "no", meta.ab = NULL)
# 这个函数有很多参数,请查阅 "icamp.big" 的帮助文档以了解更多信息。
# 输出文件:
# Test.iCAMP.detail.rda:以 R 数据格式保存的 "icres" 对象。它是一个列表对象。
#第一个元素 bNRIiRCa 是每对比较(每次替代)的组装过程相对重要性的结果。
#第二个元素 "detail" 包括每个分类单元的分类信息(名为 taxabin)、
#每个分类单元中的系统发育和分类度量结果(名为 bNRIi、RCa 等)、
#每个分类单元的相对丰度(bin.weight)、
#每个社区之间每个过程的相对重要性(processes)、
#输入设置(setting)和输入社区数据矩阵(comm)。
#有关详细信息,请参阅函数 icamp.big 的帮助文档
!!!bin.size.limit = 5该参数要根据上一步的结果进行选择。
!!!提供的代码中有其他的iCAMP分析方法(9.2-9.6),应对的不同需求,可根据需求选择。
iCAMP bin 水平统计
# 10 # iCAMP bin 水平统计
icbin=iCAMP::icamp.bins(icamp.detail = icres$detail,treat = treat,
clas=clas,silent=FALSE, boot = TRUE,
rand.time = rand.time,between.group = TRUE)
save(icbin,file = paste0(prefix,".iCAMP.Summary.rda")) # 仅用于归档结果。rda 文件会自动压缩,方便加载到 R 中。
write.csv(icbin$Pt,file = paste0(prefix,".ProcessImportance_EachGroup.csv"),row.names = FALSE)
write.csv(icbin$Ptk,file = paste0(prefix,".ProcessImportance_EachBin_EachGroup.csv"),row.names = FALSE)
write.csv(icbin$Ptuv,file = paste0(prefix,".ProcessImportance_EachTurnover.csv"),row.names = FALSE)
write.csv(icbin$BPtk,file = paste0(prefix,".BinContributeToProcess_EachGroup.csv"),row.names = FALSE)
write.csv(data.frame(ID=rownames(icbin$Class.Bin),icbin$Class.Bin,stringsAsFactors = FALSE),
file = paste0(prefix,".Taxon_Bin.csv"),row.names = FALSE)
write.csv(icbin$Bin.TopClass,file = paste0(prefix,".Bin_TopTaxon.csv"),row.names = FALSE)
Bootstrapping 测试
# 11 # Bootstrapping 测试
# 请指定处理信息表中的哪一列。
i=1
treat.use=treat[,i,drop=FALSE]
icamp.result=icres$CbMPDiCBraya
icboot=iCAMP::icamp.boot(icamp.result = icamp.result,treat = treat.use,rand.time = rand.time,
compare = TRUE,silent = FALSE,between.group = TRUE,ST.estimation = TRUE)
save(icboot,file=paste0(prefix,".iCAMP.Boot.",colnames(treat)[i],".rda"))
write.csv(icboot$summary,file = paste0(prefix,".iCAMP.BootSummary.",colnames(treat)[i],".csv"),row.names = FALSE)
write.csv(icboot$compare,file = paste0(prefix,".iCAMP.Compare.",colnames(treat)[i],".csv"),row.names = FALSE)4.3、输出结果
!输出文件:
Test.iCAMP.Boot.Management.rda: "icboot" 保存为 R 数据格式。有关对象中每个元素的描述,请参阅 icamp.boot 函数的帮助文档。
Test.BootSummary.Management.csv: 汇总启动测试结果的表格。有关输出元素 "summary" 的描述,请参阅 icamp.boot 函数的帮助文档。
Test.Compare.Management.csv: 汇总每两个组之间比较指数、效应量和显著性的表格。有关输出元素 "compare" 的描述,请参阅 icamp.boot 函数的帮助文档。
!!随后可根据输出文件内容绘制响应的图片,柱状图或者饼图或者其他图片。
5、参考文献
[1] Ning, D., Yuan, M., Wu, L. et al. A quantitative framework reveals ecological drivers of grassland microbial community assembly in response to warming. Nat Commun 11, 4717 (2020).
[2] Ning, D., Wang, Y., Fan, Y. et al. Environmental stress mediates groundwater microbial community assembly. Nat Microbiol 9, 490–501 (2024).
[3] Lu, Q., Liu, Y., Zhao, J. and Yao, M. (2024) Successive accumulation of biotic assemblages at a fine spatial scale along glacier-fed waters. iScience 27(4).
6、相关信息
!!!有需要的小伙伴可以去评论区自取今天的测试代码和实例数据。
📌示例代码中提供了数据和代码,已经小编测试,可直接运行。
⚠️本账号会不时更新代码,代码失效可联系小编~
以上就是iCAMP包进行群落构建分析全部内容。

















 2691
2691

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









