







简介
Bowtie 2是一种用于长参考序列对齐测序读取的工具。它尤其擅长将大约50-100多个字符的比对到相对较长的(例如哺乳动物)基因组。Bowtie 2支持gapped, local,and paired-end alignment模式。可以同时使用多个处理器以实现更高的对齐速度。
使用方法
bowtie2[options]* -x <bt2-idx> {-1 <m1> -2 <m2> | -U <r> |--interleaved <i> | --sra-acc <acc> | b <bam>} -S[<sam>]-x <bt2-idx> 参考基因组索引的名称。注意,这个名称不包括尾端的的.1.bt2 / .rev.1.bt2 / 等。bowtie2 首先在当前目录中查找指定的索引,如果没有找到的话,再在BOWTIE2_INDEXES 环境变量中查找。
-1 <m1> Comma-separated list of files containing mate 1s,例如 -1 flyA_1.fq,flyB_1.fq。 使用此选项指定的序列必须与 <m2> 中指定的文件对文件和读取对读取相对应。
-2 <m2> Comma-separated list of files containing mate 2s,例如 -2 flyA_2.fq,flyB_2.fq。 使用此选项指定的序列必须与 <m1> 中指定的文件对文件和读取对读取相对应。
-U <r> Comma-separated list of files containingunpaired reads to be aligned,例如lane1.fq,lane2.fq,lane3.fq,lane4.fq。
--interleaved读取interleaved的FASTQ 文件,其中前两条记录(8 行)代表配对。
输出:
--un <path> 读出没有的匹配上的 reads,并把它写入path中的文件
--al <path> 读出至少匹配一次的 reads,并把它写入path中的文件
举例
如果我有一些序列,想要去除掉这些序列中的污染
我把我的污染序列放入euk.fa这个文件
首先通过bowtie2-build 构建基因组索引
bowtie2-buildeuk.fa euk之后 使用bowtie2去除污染
bowtie2-p 40 -N 1 -x ./ euk -f <我的需要去除污染的序列> -S output.sam --un ./没有的匹配上的序列.fasta --al ./匹配上的序列.fasta-p表示使用的线程数目
-N Setsthe number of mismatches to allowed in a seed alignment during multiseedalignment。 可以设置为 0 或 1。将此设置得更高会使对齐变慢(通常慢得多)但会增加灵敏度。 默认值:0。
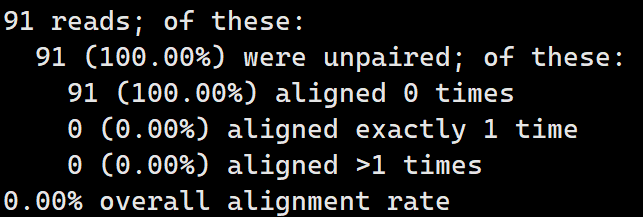
跑的时候会出现如下提示(这里意味着我的序列里边没有这个污染):
参考链接:https://bowtie-bio.sourceforge.net/bowtie2/manual.shtml#output-options















 6802
6802











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









