






最近遇到一个计算TE插入时间的需求,翻了翻网上的帖子只有 @weixin_46530443 一位大佬写过计算方式。可惜大佬在某些地方写的不够详细,前后折腾两天才算出来。为此,我将在这个帖子中详细记录LTR插入时间的计算方法,希望后来者能少走些弯路。
计算原理大佬已经写的很清楚了不在赘述:计算反转录转座子插入时间一:计算原理
一、TE注释:
1.1 EDTA安装:
可以参考另一位大佬:EDTA 最简易安装方法
但是大佬的方法对我有些不适用,我的安装方法如下(具体含义不太懂,反正装上了):
git config --global --unset http.proxy
git config --global --unset https.proxy
git pull --depth 1
git config --global http.postBuffer 524288000
git clone https://github.com/oushujun/EDTA.git
cd EDTA
conda env create -f EDTA.yml1.2 TE注释:
实际上公司给的结果中包含TE注释结果,但是由于没有指出LTR具体位置,因此采用EDTA注释,具体步骤如下:
#进入刚刚安装好的虚拟环境
conda activate EDTA
# --genome输入基因组序列文件,-cds输入CDS序列文件(可选) -t 为线程数
nohup perl EDTA/EDTA.pl --genome genome.fa -species others -cds evm_cds.fa -step all -t 20 &
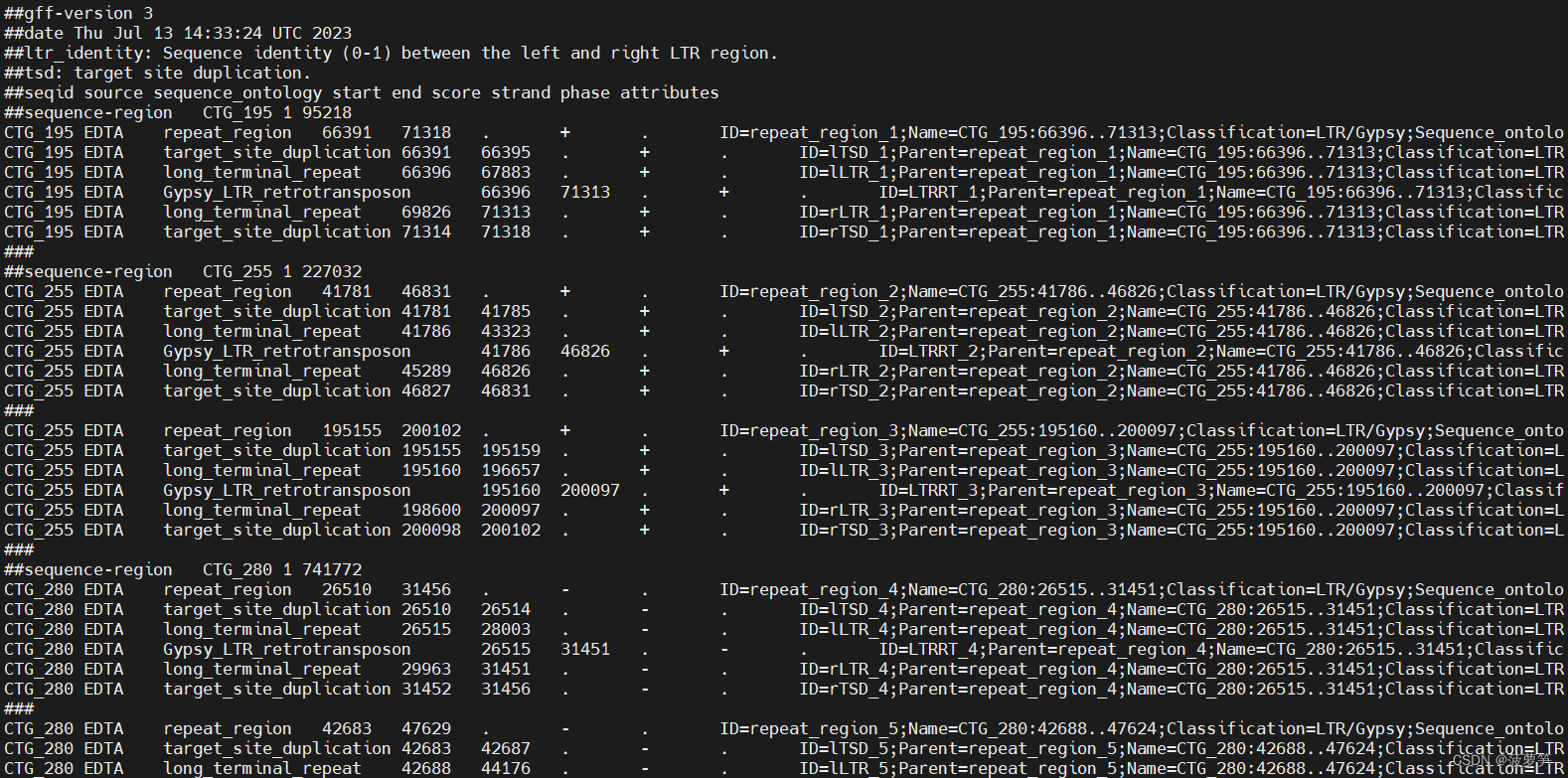
1.3 结果示例:
在EDTA的输出结果中存在一个genome.fa.mod.EDTA.raw文件夹,其中有个genome.fa.mod.LTR.intact.gff3 文件中有我们需要的信息,如下所示。每个block被“###”隔开,其中只有long_terminal_repeat是我们需要的。
二、LTR序列提取
2.1 提取5‘/3’LTR信息
为了方便后续操作这里先将5‘/3’LTR信息从总LTR注释文件中拆分出来
#从总LTR文件中提取5‘/3’LTR信息
awk '$3 == "long_terminal_repeat"{print $0}' genome.fa.mod.LTR.intact.gff3 > ../5-3LTR.gff
#拆分5‘/3’LTR
grep lLTR 5-3LTR.gff > lLTR.gff
grep rLTR 5-3LTR.gff > rLTR.gff2.2 提取LTR序列
虽然EDTA的输出结果包含了LTR的序列文件genome.fa.mod.LTR.intact.fa,但是我搞不清楚怎么从这个序列中拆出5‘/3’LTR,因此这里还是使用原始的基因组序列进行提取。运行一下Python脚本:
#coding UTF-8
# 读取fasta文件,并储存为字典,字典key为染色体名,value为序列
def read_fasta(file):
seq_list = {}
with open(file, 'r') as f:
for line in f:
if line.startswith('>'):
seq_id = line.strip()[1:].split('#')[0]
seq_list[seq_id] = ''
else:
seq_list[seq_id] += line.strip()
return seq_list
def read_gff(file): 读取GFF文件,gff_list是一个嵌套列表,每条注释信息用列表的一个元素表示,每个元素均为一个包含染色体名、起始位置、终止位置
gff_list = []
with open(file, 'r') as f:
for line in f:
gff_list.append([line.split('\t')[8].split(';')[2][5:].split(':')[0],line.split('\t')[3],line.split('\t')[4]])
return gff_list
#计算序列截取
def seq_intercept(seq_list,gff_list,LTR):
#print(seq_list.keys())
with open(f'{LTR}.fa', 'w') as f:
for seq_n in range(len(gff_list)):
#print(gff_list[seq_n][0])
#print(int(gff_list[seq_n][1])-int(gff_list[seq_n][3]))
#print(len(seq_list[gff_list[seq_n][0]][int(gff_list[seq_n][1])-int(gff_list[seq_n][3]):int(gff_list[seq_n][2])-int(gff_list[seq_n][3])+1]))
#print(f'chr:{gff_list[0]}\tSTART:{int(gff_list[seq_n][1])}\tEND:{int(gff_list[seq_n][2])}\tlen1:{int(gff_list[seq_n][2])-int(gff_list[seq_n][1])}\tlen2:{len(seq_list[gff_list[seq_n][0]][int(gff_list[seq_n][1])-1:int(gff_list[seq_n][2])])}')
f.write(f'>{LTR}:{seq_n}:\n{seq_list[gff_list[seq_n][0]][int(gff_list[seq_n][1])-1:int(gff_list[seq_n][2])]}\n')
####很可能这里是你唯一需要修改的地方####
# 主程序
if __name__ == '__main__':
cds_file = 'genome.fa'
lLTR = 'lLTR.gff'
rLTR = 'rLTR.gff'
seq_dict = read_fasta(cds_file)
lgff_list = read_gff(lLTR)
seq_intercept(seq_dict,lgff_list,"lLTR")
rgff_list = read_gff(rLTR)
seq_intercept(seq_dict,rgff_list,"rLTR")
print("END")
运行得到rLTR.fa与lLTR.fa两个序列文件,现在需要将来自同一个LTR的两端序列两两合并到一个文件中(这里会将LTR重命名,但是不影响最终结果,有需求的可以自行改进):
#创建一个ltr文件夹用于储存序列文件
mkdir ltr/
#这里的16038是 lLTR.fa和 rLTR.fa的行数,用wc -l查看
for ((i=0;i<16038;i+=2)); do head -n $i lLTR.fa |tail -n 2 >ltr/ltr$i.fa;head -n $i rLTR.fa |tail -n 2 >>ltr/ltr$i.fa; done
三、MEGA计算K值
这部分是卡了我最长时间的。。。
3.1 MEGACC安装
我在这里安装的是Linux版本,windows应该也可以
参考文章:如何使用Mega cc
前往MAGA官网下载Linux版MAGA:http://www.megasoftware.net/megaccusage.php 并上传至服务器
#解压就可以使用了:
tar -zxvf megacc_11.0.13_amd64.tar.gz3.2 使用MEGACC进行序列比对
这里生成配置文件可以参考:计算反转录转座子插入时间三:MEGA批量化处理
这里是我和大佬最不一样的地方。用了大佬的方法后一直报错,起初以为是windows版不行后转战Linux,结果还是报错,最后在Linux上用for循环的方法完成了(window下应该也可以,但是我没有尝试)。
在个人电脑中打开MEGA,点击右下角PROTOTYPE,进入模拟模式,选择序列类型(我使用Nucleotide non-coding)ALIGN——MUSCLE,使用默认参数点击确定生成比对配置文件.mao。
#生成文件列表:
ls ltr/*fa >filelist.txt
#生成MEGA比对文件输出文件夹
mkdir output
#循环运行mega,此处与大佬文章中不同,之前学大佬的方法疯狂报错,但是这样就可以了
for i in `cat filelist.txt` ;do megacc -a muscle_align_nucleotide.mao -d $i -o output/ ;done
输出有很多,需要的是.meg文件,用MEGA打开长这样:
3.3 使用MEGACC进行K值计算
与3.2的方法类似,这里也采用的for循环,此处的配置文件自行参考上方的文章:
#配置文件distance_estimation_pairwise_nucleotide.mao自行参考大佬文章生成
#创建K值输出文件夹
mkdir output2
#创建文件列表
ls output/*.meg > megfilelist.txt
#循环计算K值
for i in `cat megfilelist.txt` ;do megacc -a distance_estimation_pairwise_nucleotide.mao -d $i -o outputa2/ ;done这样就得到了K值计算结果,虽然以.meg结尾,但是MEGA无法打开。用文本编辑器查看:
#mega
!Title: ;
!Format DataType=Distance DataFormat=LowerLeft NTaxa=2;
!Description
Using the following analysis options:
No. of Taxa=2
Analysis=Distance Estimation
Scope=Pairs of taxa
Variance Estimation Method=None
No. of Bootstrap Replications=Not Applicable
Substitutions Type=Nucleotide
Model/Method=Kimura 2-parameter model
Substitutions to Include=d: Transitions + Transversions
Rates among Sites=Gamma Distributed (G)
Gamma Parameter=1.00
Pattern among Lineages=Same (Homogeneous)
Gaps/Missing Data Treatment=Complete deletion
Site Coverage Cutoff (%)=Not Applicable
Has Time Limit=False
Maximum Execution Time=-1
datatype=snNucleotide
containsCodingNuc=False
MissingBaseSymbol=?
IdenticalBaseSymbol=.
GapSymbol=-
Labelled Sites=All Sites
No. of Sites=1489
d=Estimate
;[1] #lLTR:4:
[2] #rLTR:4:[ 1 2 ]
[1]
[2] 0.00067227
最下方的 0.00067227就是我们需要的K值。
四、计算插入时间
这里也直接套用大佬写的python脚本,但是我对其做了一些改进——删除了多余的代码以及做了一些注释:
import os
import sys
#获取K值文件列表
def getfile(fold):
for names in os.walk(fold):
file_list = []
for filename in names:
if type(filename) == str:
path = filename
if type(filename) == list:
if filename != []:
for i in filename:
if i.endswith("meg"):
new_filename = path + i
file_list.append(new_filename)
return file_list
#计算插入时间,这里需要注意碱基突变率
def gettime(file):
with open(file, "r") as fi:
ltr_pair = ""
ltr_time_dic = {}
ltr_dic = {}
f = fi.readlines()
for line in f:
lin = line.strip().split(" ")
if line.startswith("[1]"):
if len(lin) == 2:
ltr1 = lin[1].strip("#")
if line.startswith("[2]"):
if len(lin) == 2:
ltr2 = lin[1].strip("#")
ltr_pair = ltr1 + ";" + ltr2
ltr_dic[ltr1] = ltr2
if len(lin) == 3:
distance = float(lin[2])
#####这里的r2是碱基突变率#####
r2 = 1.99*10**(-9)
time = (distance/(2*r2))/1000000
ltr_time_dic[ltr_pair] = time
out_line = ltr_pair + "\t" + str(time)
#print(ltr_pair, time)
#print(time, ltr_time_dic)
return out_line ###ltr_time_dic
#写入文件
def writelis(lis, fil):
with open(fil, "w") as out_f:
for it in lis:
line1 = it + "\n"
out_f.write(line1)
out_f.close()
#主程序
f_list = getfile(r"output2/") #这里需要根据你自己设置的文件夹决定
new_list = []
for item in f_list:
time_line = gettime(item)
new_list.append(time_line)
writelis(new_list, "ltr_insertime_out.txt") #这里可以修改你的输出文件名称
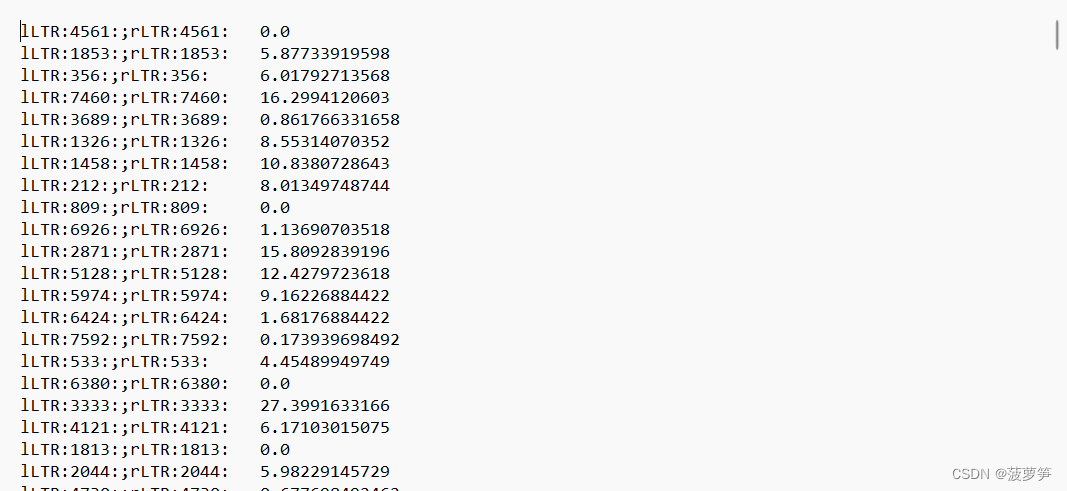
运行以上Python代码可以得到以下文件,第一列是LTR名称(重命名后),第二列是插入时间(百万年):
五、绘制密度图:
这里使用R语言:
library(ggplot2) #导入ggplot2包
time <- read.table("ltr_insertime_out.txt") #读取文件
den <- density(time$V2) #计算时间密度
denx=den$x #保存X轴
deny=den$y #保存Y轴
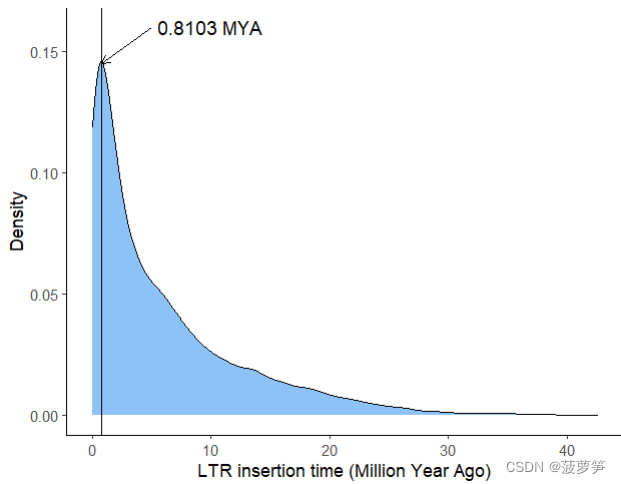
#denx[which.max(deny)]就是最大密度所在年份
p <-
ggplot()+
geom_density(mapping=aes(x=time$V2),fill="dodgerblue2",alpha=0.5)+
geom_vline(xintercept = denx[which.max(deny)])+
#下面几行根据具体情况修改
geom_segment(mapping=aes(xend=denx[which.max(deny)],yend=0.145,x=5,y=0.16),arrow = arrow(length=unit(0.2, "cm")))+
annotate("text", x = 10, y = 0.16,label = paste(round(denx[which.max(deny)],4),"MYA"),size=4)+
labs(x="LTR insertion time (Million Year Ago)",y="Density")+
xlim(c(0,15))+
theme_classic()
ggsave("LTR_insert_time.pdf",p,width = 210*0.4 , height = 210*0.4,units="mm")
输出结果如下:
OK,大功告成














 6143
6143











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









