






 DAS_Tool是一种用于宏基因组学的dereplication, aggregation和scoring策略,旨在整合不同binning方法的结果,提高bins的质量和完整度。该工具通过预测和评分单拷贝基因,迭代选择高分bins,去除冗余,同时展示strain variation。输入包括多个binning方法的scaffolds2bin文件,contig的fasta文件,输出包括非冗余的高分bins及其评估信息。DAS_Tool支持多种搜索引擎和参数调整,可用于优化宏基因组数据的分析结果。
DAS_Tool是一种用于宏基因组学的dereplication, aggregation和scoring策略,旨在整合不同binning方法的结果,提高bins的质量和完整度。该工具通过预测和评分单拷贝基因,迭代选择高分bins,去除冗余,同时展示strain variation。输入包括多个binning方法的scaffolds2bin文件,contig的fasta文件,输出包括非冗余的高分bins及其评估信息。DAS_Tool支持多种搜索引擎和参数调整,可用于优化宏基因组数据的分析结果。
参考https://github.com/cmks/DAS_Tool
DAS: dereplication, aggregation and scoring strategy
DAS Tool可以将不同宏基因组分箱后得到的bins进行整合,得到更多高质量,高完整度,非冗余的bins,还能更好地展示strain variation微生物株系之间的差异。
在对DAS Tool进行input时,可以选用尽可能多的binning方法的结果,即使是一些只获得很少高质量的bins的binning方法也可能获得一些其他方法忽略的bins。
ABAWACA performs a hierarchical clustering on tetranucleotide frequencies and differential coverage, and takes marker genes into account. CONCOCT uses Gaussian mixture models and tetranucleotides frequencies with differential coverage9 . MaxBin 2 is based on an expectation-maximization algorithm and uses tetranucleotides, differential coverage and marker genes13. MetaBAT applies a k-medoid clustering on tetranucleotide frequencies and differential coverage.(引用自Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy | Nature Microbiology)
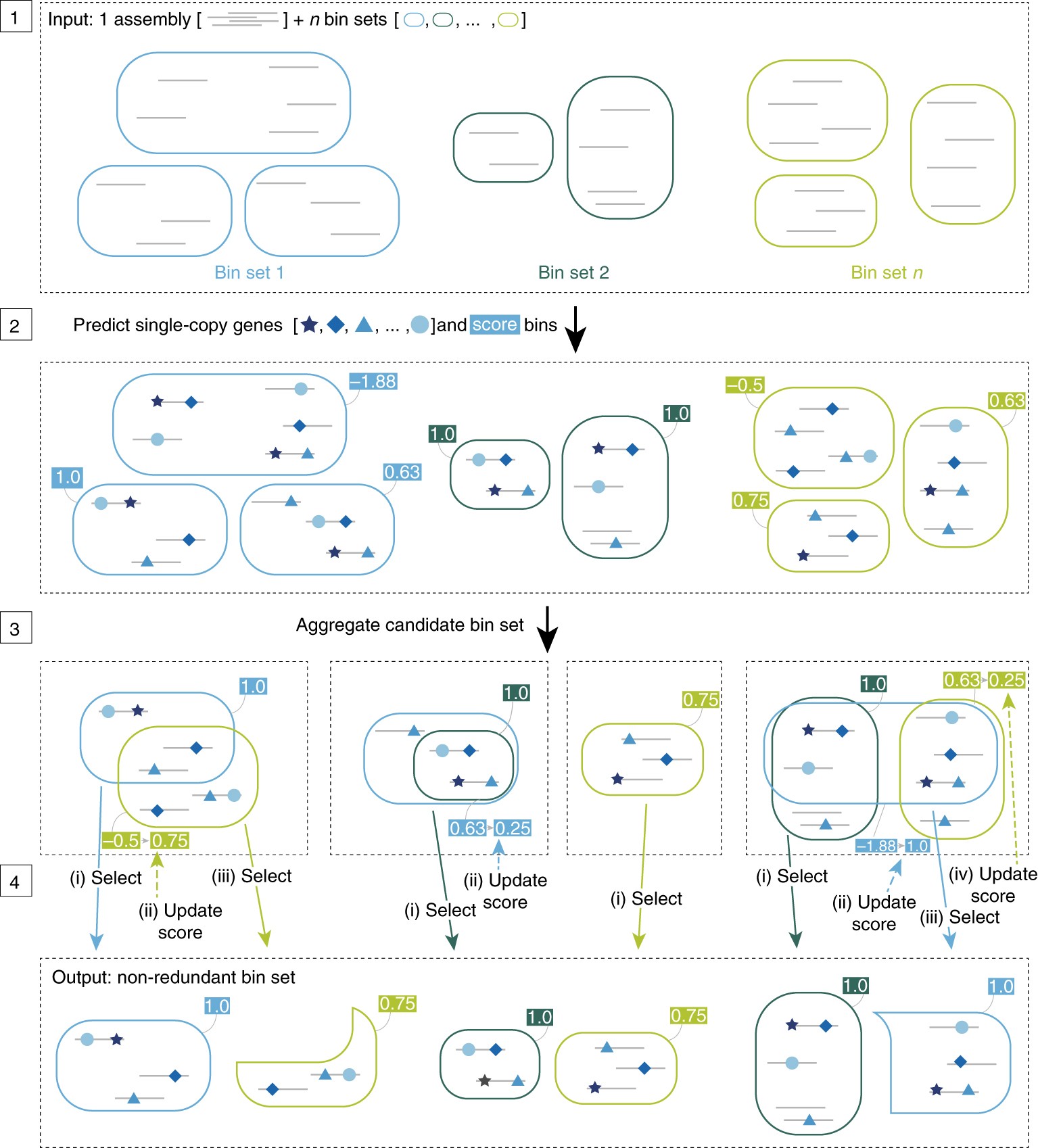
DAS的核心思想是进行基于单拷贝基因评分判断bin质量的迭代。
步骤1:DAS工具的输入文件包括拼接结果中的scaffolds序列(灰线表示)和来自不同binning工具得到的bins集合(相同颜色的圆角矩形表示由同一binning方法得到的bins);
步骤2:预测每个bins中scaffold的单拷贝基因(蓝色形状表示),并进行打分;
步骤3:在所有结果中,将相同的bins进行合并,作为这个bins的备选集合;
步骤4:迭代选择高分bins,并更新集合剩余部分候选bins的分数。如果有分数相同的情况,选择scaffold N50值更高的bin。 N50值:覆盖50%基因组所需要的最小的contig长度
测序得到若干条reads,这些reads进行拼接,如果完全可以拼接起来,中间没有gap的序列称为contig,即连续的意思。如果中间有gap,但是可以知道gap的长度,这样的序列就叫做scaffold, 即脚手架(非连续)的意思。把contig 和 scaffold 从长到短进行排列,然后相加,当恰好加到1M的50%,也就是500k的时候 ,那一条 contig 或者scaffold 的长度就叫做Contig N50和Scaffold N50。很明显这个数值越大说明组装的质量越好。
即:从最长的开始倒数,数到长度为总长度一半的片段,最后一个被数到的片段越长,说明长的片段越多,最后组装的质量越好。
引用自基因组测序中N50和N90到底指什么?_Mr番茄蛋的博客-CSDN博客_n50是什么意思
最终输出包括来自不同输入文件预测的非冗余高分bins(分数大于threshold t)。
CheckM首先基于完整的已测序细菌基因组作为参考基因组,构建基因组的进化树,构建每个谱系(可以理解为一类物种)的单拷贝基因集(single copy genes,SCGs,为什么是单拷贝?因为这样可以开展基因组混合程度、污染程度等的评估)。在使用时,将Bin与参考基因组一起建树,基于进化关系找到Bin的参考物种,然后结合参考物种的单拷贝基因集,计算两个重要指标。Completeness,完整度,Bin基因与对应SCGs相比,基因数量是否完整,取值[0,100%],数值越大,表示Bin质量越好;Contamination,污染度,Bin基因包含多个物种的SCGs,即一个Bin存在多个物种的程度,取值[0,100%],数值越小,表示Bin质量越好。
实操
DAS_Tool -i methodA.scaffolds2bin,...,methodN.scaffolds2bin
-l methodA,...,methodN -c contigs.fa -o myOutput
-i, --bins Comma separated list of tab separated scaffolds to bin tables.
-c, --contigs Contigs in fasta format.
-o, --outputbasename Basename of output files.
-l, --labels Comma separated list of binning prediction names. (optional)
--search_engine Engine used for single copy gene identification [blast/diamond/usearch].
(default: usearch)
--write_bin_evals Write evaluation for each input bin set [0/1]. (default: 1)
--create_plots Create binning performance plots [0/1]. (default: 1)
--write_bins Export bins as fasta files [0/1]. (default: 0)
--proteins Predicted proteins in prodigal fasta format (>scaffoldID_geneNo).
Gene prediction step will be skipped if given. (optional)
--score_threshold Score threshold until selection algorithm will keep selecting bins [0..1].
(default: 0.5)
--duplicate_penalty Penalty for duplicate single copy genes per bin (weight b).
Only change if you know what you're doing. [0..3]
(default: 0.6)
--megabin_penalty Penalty for megabins (weight c). Only change if you know what you're doing. [0..3]
(default: 0.5)
--db_directory Directory of single copy gene database. (default: install_dir/db)
--resume Use existing predicted single copy gene files from a previous run [0/1]. (default: 0)
--debug Write debug information to log file.
-t, --threads Number of threads to use. (default: 1)
-v, --version Print version number and exit.
-h, --help Show this message.
-i 输入不同binning方法得到的bins结果,文件格式为tabular scaffolds2bin file,包括tab分隔开的scaffold-IDs和bin-ID。
Scaffold_1 bin.01
Scaffold_8 bin.01
Scaffold_42 bin.02
Scaffold_49 bin.03-l 与-i input的文件中一一对应的binning方法,逗号隔开
-c 组装好的contig的fasta文件
-o 文件输出至指定文件夹,输出文件包括DASTool_summary.txt(输出的bins和其质量与完整性的估计);DASTool_scaffolds2bin.txt(输出的bins和其对应包含的scaffold)
--search_engine单拷贝基因识别的搜索方法,默认为usearch(需预先安装),还包括blast和diamond
--write_bin_evals对每一个输入的bin set进行评估([method].eval)
--write_bins以fasta文件输出bins (DASTool_bins)
--proteinsProdigal预测的蛋白的fasta格式
--score_threshold选择bin的阈值
--create_plots显示每种方法的高质量 bin 的分布(DASTool_hqBins.pdf, DASTool_scores.pdf)
安装
可以使用bioconda进行安装,预先安装miniconda。
先添加conda channel
conda config --add channels defaults
conda config --add channels bioconda
conda config --add channels conda-forge再使用conda进行安装
conda install -c bioconda das_tool格式转换
输入的bins结果文件应该为tabular scaffolds2bin文件格式,可使用下面代码进行转换
src/Fasta_to_Scaffolds2Bin.sh
参数:
-i 需要转换的文件的路径
-e fasta > my_scaffolds2bin.tsv-e 文件的扩展名

















 7684
7684

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









