








–https://doi.org/10.1101/2024.01.25.577152
scMulan: a multitask generative pre-trained language model for single-cell analysis
scMulan是清华-张学工团队的研究成果,模型架构基于transformer的decoder,做成了生成式模型。
模型代码详情:https://github.com/SuperBianC/scMulan/tree/main
留意更多内容,欢迎关注微信公众号:组学之心
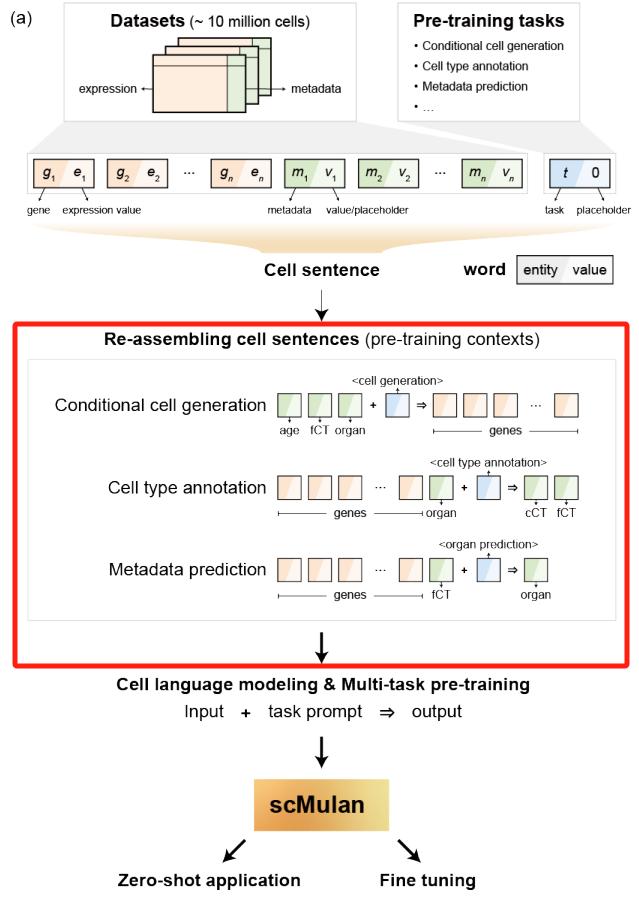
用于预训练的数据
从hECA数据库中选择一个子集来训练,命名为 hECA-10M。该数据包含心脏、大脑、肺、肝脏、骨髓、血液和胸腺合计1000 多万单细胞转录组数据,并附带的meta数据记录了包括器官来源、器官内的特定区域、供体年龄、供体性别、测序技术和细胞类型在内的属性。
将细胞类型注释映射到统一人体图谱框架 (uHAF)的框架上,统一细胞类型注释。
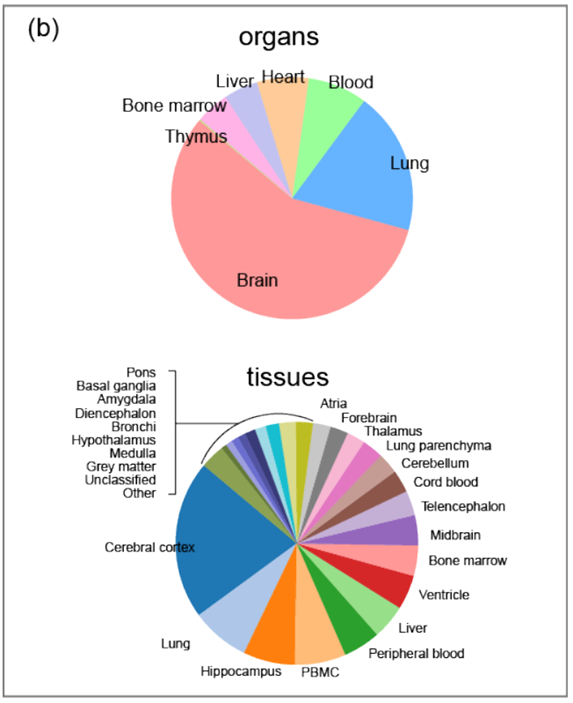
用 scanpy 包把数据归一化和 log1p 转换,并选出整个数据集中top 2,000 位的高变异基因。这 2,000 个高度可变的基因一直用作所有数据集的特征。

将这个综合数据集随机分为两部分,90% 的细胞指定用于预训练,其余保留用于验证。
1.模型架构
scMulan模型设计参考了openAI的
–https://cdn.openai.com/research-covers/language-unsupervised/language_understanding_paper.pdf

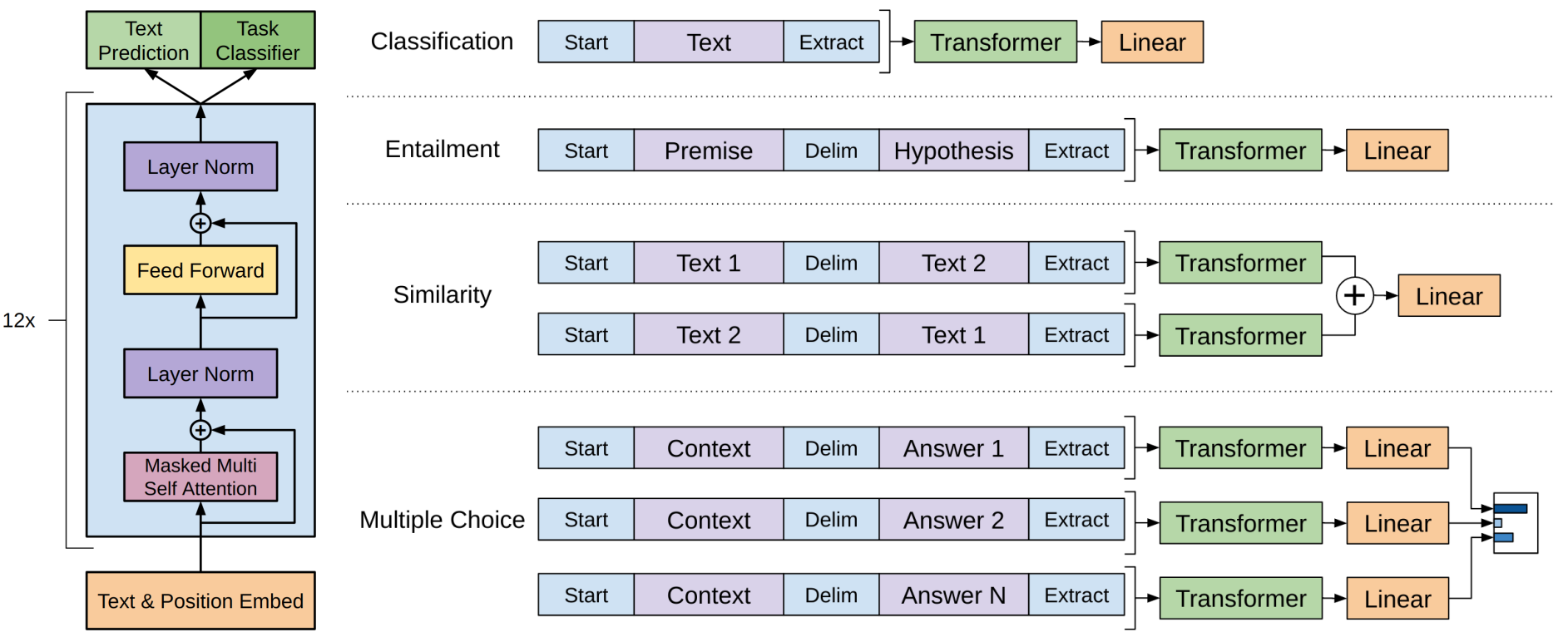
该模型类似transformer模型的decoder部分,用于对不同任务进行微调的输入转换。
-
分类任务 (Classification):输入序列经过标记的起始符号(Start),文本内容(Text),分隔符号(Delim),以及最终提取的表示(Extract),然后经过Transformer层和线性层,进行分类任务。
-
蕴含任务 (Entailment):这里输入两个文本片段:前提(Premise)和假设(Hypothesis)。两者经过Transformer层处理后,利用线性层进行蕴含关系的判别。
-
文本相似性任务 (Similarity):两段文本(Text 1和Text 2)分别通过Transformer层处理,然后将这两段表示相加(+)后再通过线性层,最终计算文本相似性。
-
多项选择任务 (Multiple Choice):这里模型处理的是多项选择任务。不同的上下文(Context)和选项(Answer N)通过Transformer后分别计算分数,并通过线性层输出最终选择。
在这些任务中,模型一般通过最小化预测下一个词的误差来学习生成整个句子:
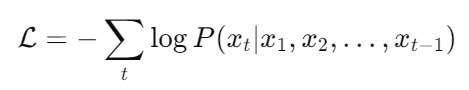
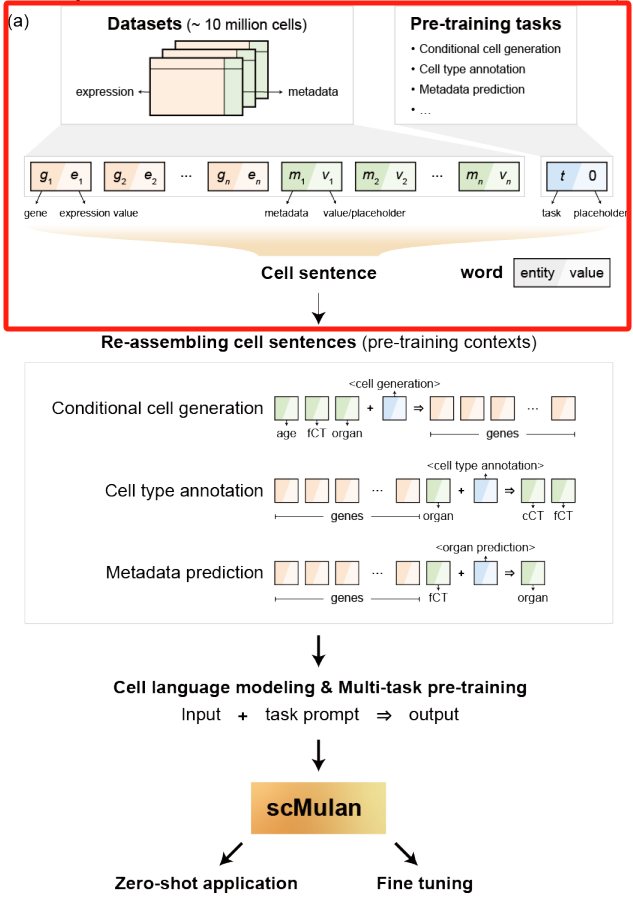
1.1 scMulan 中 cell-sentence 构建
这一步是模型输入embedding之前的操作,将每个细胞的信息概念化为一个“细胞句子”(c-sentence),其中包含所有表达基因的符号和表达值以及细胞的meta数据。
在这个框架内,将 c-sentence中的“基因词”(𝑊_𝐺)定义为基因及其相应表达水平的配对,分别称为“entity”和“value”。举个例子:表达水平为 0.2 的基因 CD3D 将表示为 𝑊_𝐺(CD3D,0.2)。
其他类别的术语如下:
- ‘word-of-metadata’ (𝑊_M) ,实体是meta数据信息中的术语;
- ‘word-of-task’ (𝑊_T) ,表示与c-sentence关联的特定任务;
- ‘word-of-special-tokens’ (𝑊_S),实体是特殊术语,例如“#E#”表示c-sentence的结尾。
这些类别用一个零值,因为它们不像基因那样具有表达水平。
比如一个心脏的心肌细胞的c-sentence示例:
(Heart 0), (cardiomyocyte cell, 0), (A2M, 0.2), (ZNF385B, 0.6), … , (CD83, 0.1), (E, 0)
这样就把一个细胞的基因表达信息和其meta信息变成了一个细胞句子c-sentence。
1.2 c-sentence的输入和输出
为了指定模型应执行的任务,需要在输入c-sentence中添加了预定义的任务prompts( W T s W_Ts WTs),它们的实体表示目标任务。
输入1:
如果我们要求模型生成心脏的“心肌细胞”数据,可以先设定基因 g 1 g_1 g1 和 g 2 g_2 g2 的表达值分别设置为 0.8 和 0.6,则输入的细胞句子是:如“(心肌细胞,0.0),(心脏,0.0),( g 1 g_1 g1,0.8),( g 2 g_2 g2,0.6),(<细胞生成>,0.0)”。
输出1:
按顺序输出“( g 3 g_3 g3, 0.3), ( g 9 g_9 g9, 0.5), …, ( g n g_n gn, 0.2), (#E#, 0.0)”这个细胞句子,其中包含此“心肌细胞”的 W G s W_Gs WGs其余部分。这里, W T W_T WT ‘<细胞生成>’ 用作表示细胞生成任务的prompt,而 ‘#E#’ 用生成细胞句子的结尾标识。
输入2:
在细胞类型注释任务中,指令通过包含任务prompts“<细胞类型注释>”的细胞句子来完成。例如输入的细胞句子是“(CD3D, 0.7), (CD4, 0.6), …, (CCR7, 0.3), (<细胞类型注释>, 0.0)”
输出2:
模型预计输出“(T细胞, 0.0), (CD4 T细胞, 0.0), (#E#, 0.0)”。
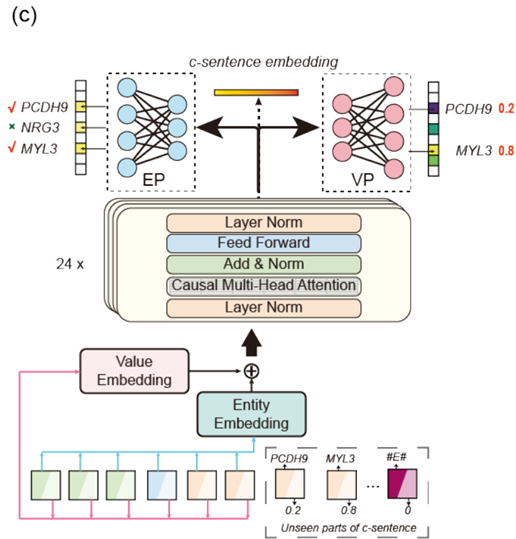
1.3 模型架构
研究基于Transformer的解码器架构,构建了scMulan,灵感来自OpenAI的GPT模型,并针对单细胞数据的特点进行了关键改进,主要涵盖了三个领域:双嵌入层、数值预测头和生成预测。
双嵌入层
用于把输入实体和数值投射为具有相同维度的高维embedding,实体embedding层( E e E_e Ee)和数值embedding层( E v E_v Ev)分别投射。然后,实体embedding和数值embedding相加,结合了基因的身份和表达水平,作为输入传递给Transformer层。
数值预测头(VP)
为了生成带有数值的输出,研究引入了数值预测头(VP)。VP通过Transformer的最后一层中的logits来预测基因在 W G s W_Gs WGs中的表达水平:
生成预测
单细胞数据中的基因在本质上位置信息不重要,因此移除了位置编码,保留了自注意力机制的位置信息不敏感性。自注意力机制的特点是能够让模型在没有位置信息的情况下,依然能够在输入的不同部分之间捕捉到复杂的相互关系。
在生成任务中,模型要预测下一个步骤的输出。scMulan 基于给定的输入词预测每个时间步中的所有可能的实体和对应的数值。
在推理阶段,模型从实体预测头的 logits 分布中抽样,逐步生成每个实体。模型会根据预测的概率分布,从多个可能的实体中选择一个进行生成。这是一种自回归的生成方式,每一步生成的实体依赖于之前的预测。
虽然输入序列仍然依赖于 c-sentence 的位置(即 c-sentence 中基因表达值的顺序),但为了减少输入顺序对模型的影响,研究采用了随机打乱 c-sentence 中基因的策略。通过打乱基因的顺序,可以减少模型对基因排列顺序的依赖,使得模型更加稳健,能够更好地处理不同排列的输入数据。
1.4 预训练任务
条件细胞生成:
作为一项基础任务,条件细胞生成涉及根据给定条件生成基因及其相应的表达值。这些条件可能包括各种因素,包括组织名称、细胞类型、供体年龄、序列技术、给定的基因实体及其表达值,或它们的任何联合组合。这项任务要求模型掌握相互关联的基因和meta数据条件的复杂关系。在训练过程中,scMulan 设置为仅对meta数据术语有条件
分层单元类型注释:
此任务是为给定细胞生成细胞类型注释的双重分层级别,其中包括细粒度和粗粒度细胞类型。目的是引导模型了解细胞类型固有的层次结构,以及它们与基因表达的联系,从而增强对细胞类型间同质性和异质性概念的看法。
其他meta数据预测:
在本研究中,我们通过器官区域预测来演示这项任务。此分类任务要求模型确定给定输入细胞的原始器官/组织和特定区域。这是一个关键功能,可在模型中灌输细胞的空间相关表达方差。由于meta数据中的信息包含细胞多样性的重要来源,因此可以以相同的方式添加细胞多样性的任何进一步协变。
1.5 细胞语言建模
传统的自然语言模型
通过按顺序乘以条件概率来优化联合概率分布:

对于一组 ( x 1 , x 2 , . . . , x n ) (x_1,x_2,... ,x_n) (x1,x2,...,xn),每个 x x x包含可变长度的符号序列 ( s 1 , s 2 , . . . , s m ) (s_1, s_2, ... ,s_m) (s1,s2,...,sm)。这些模型旨在通过估计 p ( w i ∣ w 1 , w 2 , . . . , w i − 1 ) p(w_i|w_1, w_2, ... , w_{i−1}) p(wi∣w1,w2,...,wi−1) 来最大化对数似然,从而在给定前一个符号的情况下预测下一个符号。
单细胞c-sentence的上下文
基因的顺序是非连续的,没有内在意义,因此模型的目标不是预测下一个基因,而是预测所有可能的后续基因(实体)的集合。
研究重新制定目标来估计 P ( E − { e 1 , e 2 , … , e i } | e 1 , e 2 , … , e i ) P\left( E - \{ e_1, e_2, \dots, e_i \} \middle| e_1, e_2, \dots, e_i \right) P(E−{e1,e2,…,ei}∣e1,e2,…,ei),其中 E E E 表示c-sentence中所有实体的集合, { E − { e 1 , e 2 , … , e i } } \{ E - \{ e_1, e_2, \dots, e_i \} \} {E−{e1,e2,…,ei}} 是尚未设定条件的实体集合。
最小化c-sentence的每个位置的负对数似然之和
此外,模型基于已知部分实体及其值,推断其他未知实体的值或状态。因此,目标是最小化条件概率 P ( { E − { e 1 , e 2 , … , e i } } | e 1 , e 2 , … , e i ; v 1 , v 2 , … , v i ) P\left( \{ E - \{ e_1, e_2, \dots, e_i \} \} \middle| e_1, e_2, \dots, e_i; v_1, v_2, \dots, v_i \right) P({E−{e1,e2,…,ei}}∣e1,e2,…,ei;v1,v2,…,vi),尽可能提高对剩余实体的预测精度。其中 v i v_i vi 是实体 e i e_i ei 的值。
为了有效地利用自我注意力计算,研究进一步最小化了c-sentence的每个位置的负对数似然之和 $\sum_i -\log\left( { E - { e_1, e_2, \dots, e_i } } \middle| e_1, e_2, \dots, e_i; v_1, v_2, \dots, v_i \right)
$ :
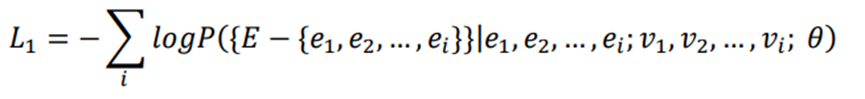
此外,研究采用回归来预测给定观察到的实体及其值的未观察到的基因的表达值,并在训练过程中把c-sentence 中的基因实体随机打乱。
对于数值预测使用均方误差(MSE):
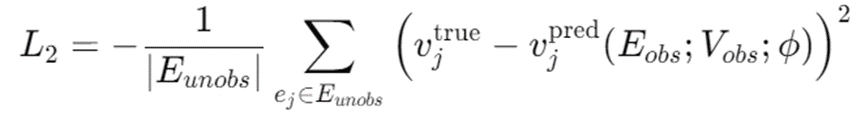
其中, v j t r u e v_j^{true} vjtrue表示实体 e j ej ej 的真实数值, v j p r e d v_j^{pred} vjpred表示在给定观察到的实体 𝐸_{𝑜𝑏𝑠}及其数值V_{𝑜𝑏𝑠}的情况下,scMulan的数值预测头给出的预测数值,参数为 𝜙,并且 𝐸_{𝑢𝑛𝑜𝑏𝑠} = {𝐸−𝐸_{𝑜𝑏𝑠}}表示未观察到的实体集合。
实体和数值预测头:
分别为 h e a d e head_e heade 和 h e a d v head_v headv,利用了共享的隐藏嵌入,这些嵌入源自多层Transformer解码器,其中 E e E_e Ee和 E v E_v Ev分别是实体和数值嵌入层,𝑛是层的数量, h e a d e head_e heade 和 h e a d v head_v headv 是多层感知机(MLP):
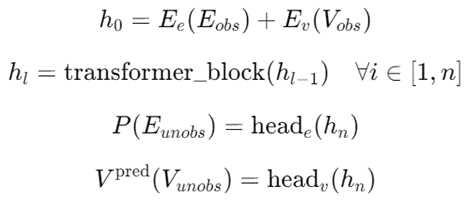
通过加权因子 𝜆 优化复合目标
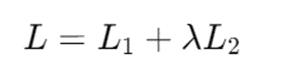
1.6 模型细节
scMulan 是一个仅使用 Transformer 解码器的模型,由 20 个 Transformer 解码器块组成,每个块有 20 个注意力头,隐藏层维度为 1120。
模型具有用于实体和数值embedding的额外embedding层,并有用于实体和数值预测的预测头。实体embedding层将基因实体作为输入并将其投射到维度为 1120 的embedding空间。数值embedding层与 scGPT 类似,scGPT 将基因的表达作为输入,将其分箱为离散级别,并将表达值投射到与实体相同的embedding维度。预测头在解码器的每个时间步预测c-sentence中的后续实体和数值。
在训练阶段,scMulan 使用自回归方法,利用上下文信息生成输出序列。模型在 PyTorch 2.0 平台上实现,结合了 Flash Attention 技术,以优化训练过程中的计算效率并提高训练速度。在 hECA-10M 数据集上预训练了 scMulan 共 245,000 步,批次大小设置为 40,梯度累计增加至 320,相当于每个批次大约处理 320,000 个词(token)。预训练过程使用了 192 小时,运行在四张 Nvidia-A800 GPU 上。
在推理阶段,scMulan 引入了 KV 缓存技术,有效地减少了推理的计算需求。
该模型支持扩展的 2k 上下文输入。scMulan 的参数总数达到 3.86 亿。
2.下游实验
2.1 无需微调即可对三个数据集进行细胞类型注释。
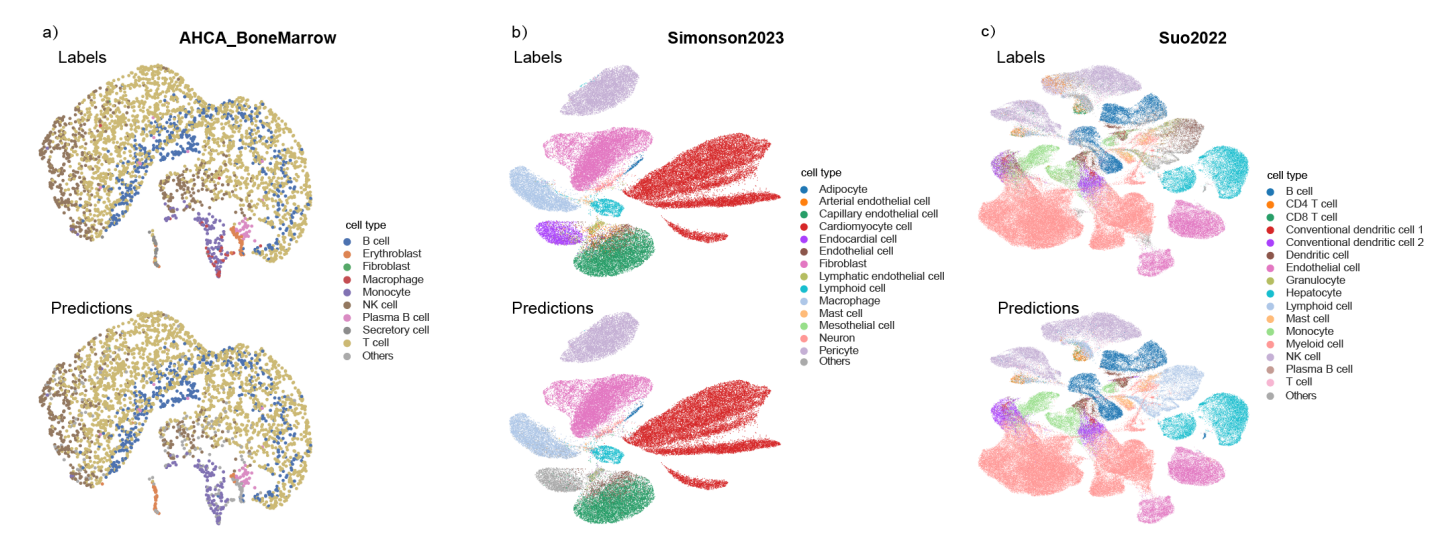
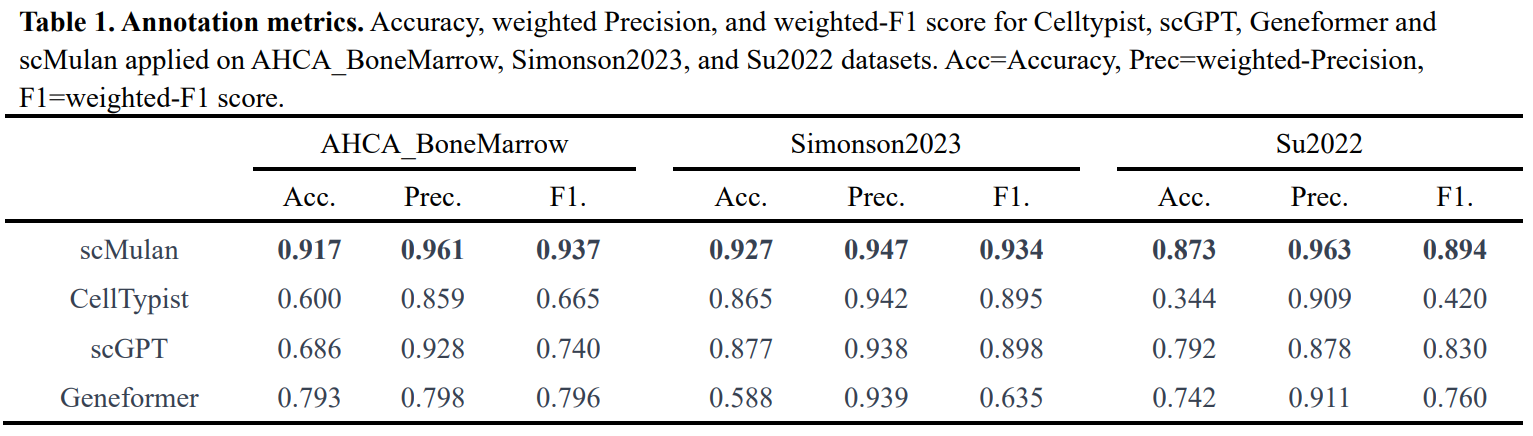
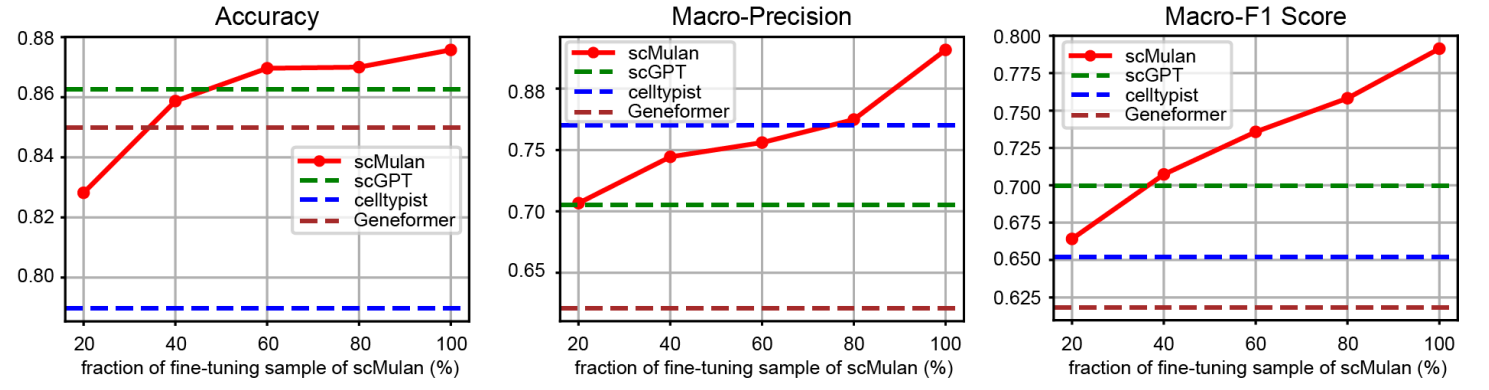
2.2 scMulan 通过微调提高了细胞类型注释的性能
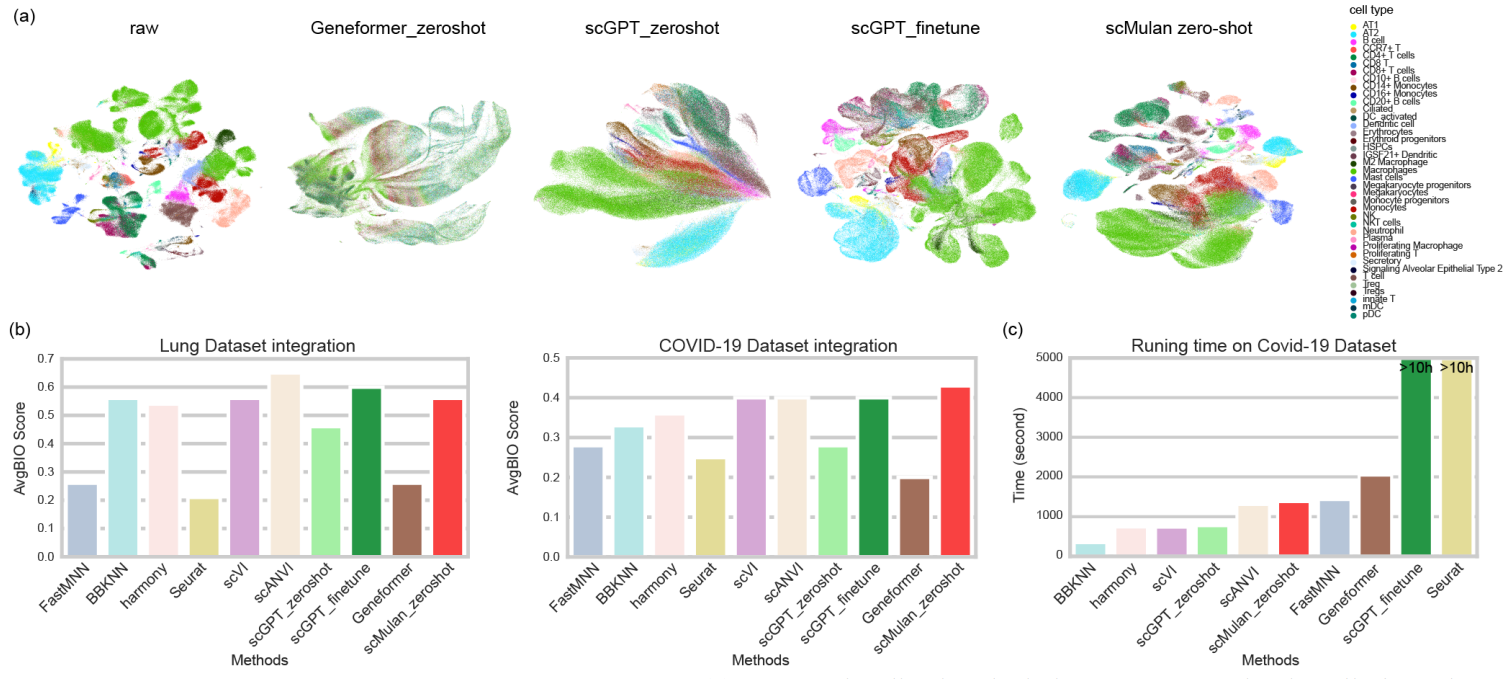
2.3 可实现跨多个批次的快速准确数据整合,无需进行微调
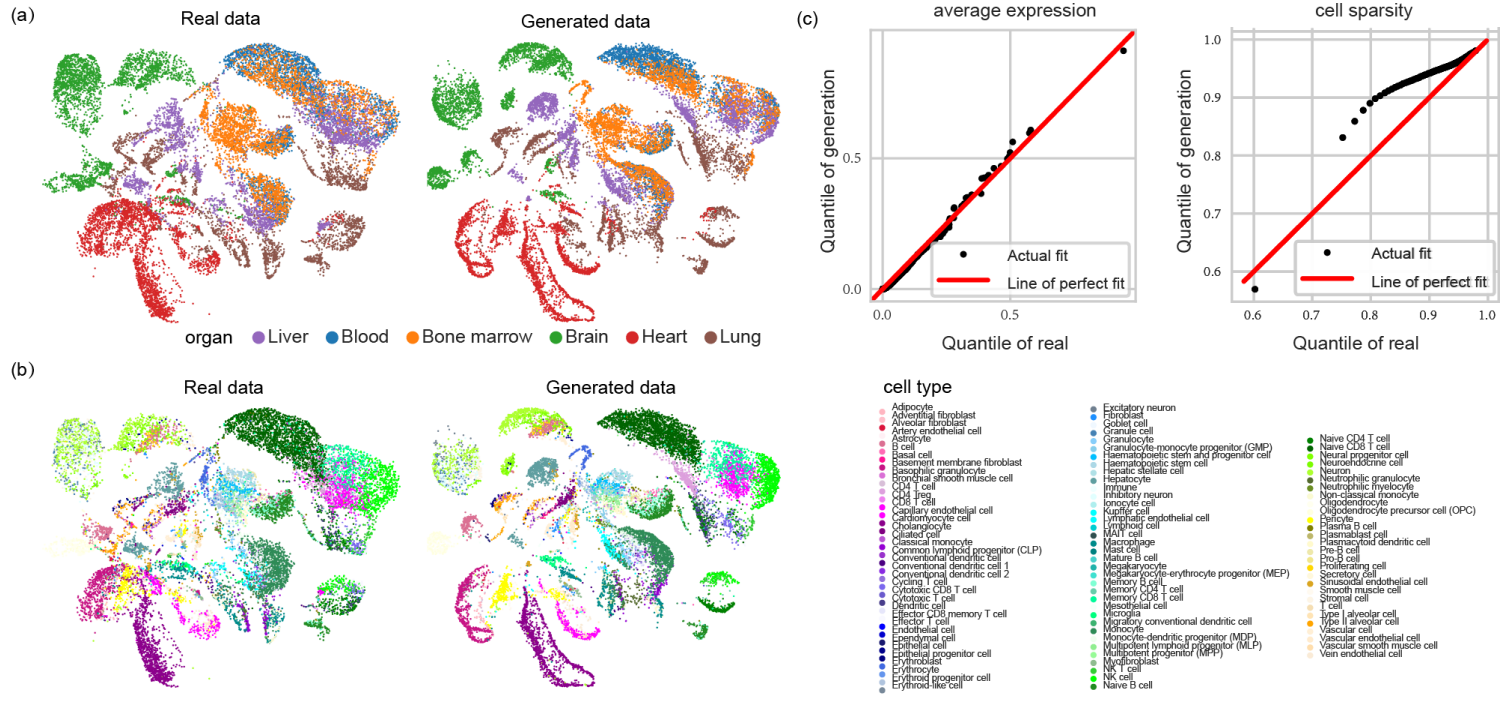
2.4 条件性生成单细胞表达谱
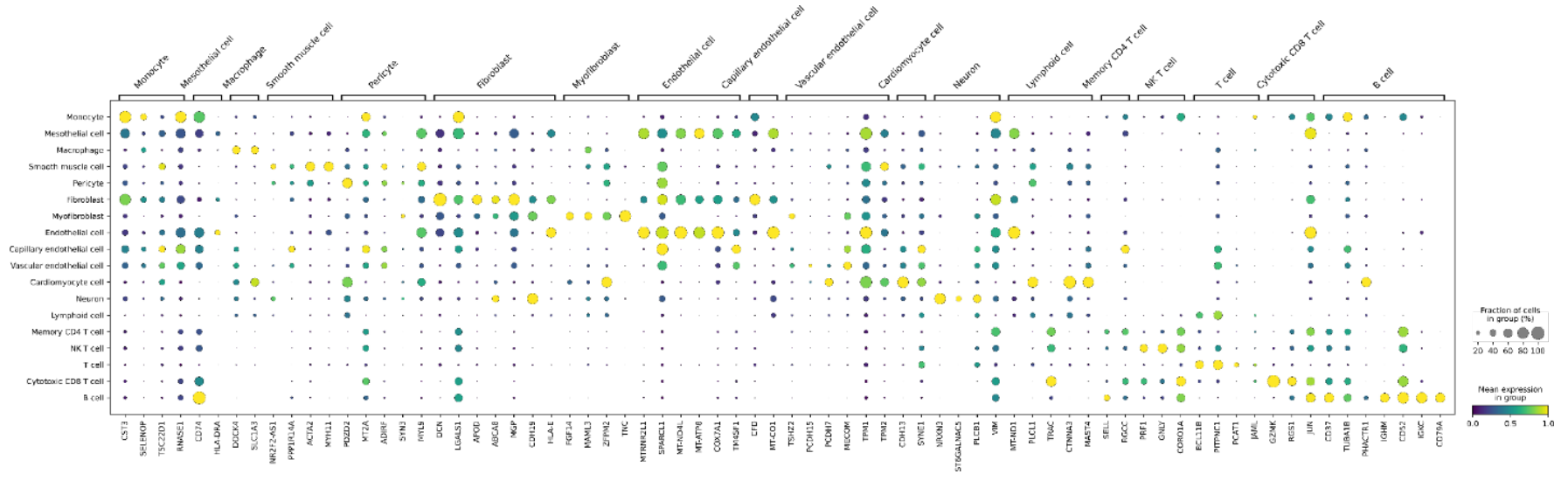
2.5 模型的可解释性
检查与细胞类型注释相关的基因来探索 scMulan 的可解释性
3.模型代码
先安装好模型
https://github.com/SuperBianC/scMulan
conda create -n scMulan python==3.10
conda activate scMulan
pip install -r requirements.txt
3.1 用预训练好的模型进行细胞注释
来源:https://github.com/SuperBianC/scMulan/blob/main/Tutorial-cell_type_annotation.ipynb
作者这里提供了肝的数据,可以从这里直接下载:https://cloud.tsinghua.edu.cn/f/45a7fd2a27e543539f59/?dl=1
预训练的权重从这里下载:https://cloud.tsinghua.edu.cn/f/2250c5df51034b2e9a85/?dl=1
1.加载h5ad数据
import os
os.environ["CUDA_VISIBLE_DEVICES"] = "0,1,2,3"
import scanpy as sc
import scMulan
from scMulan import GeneSymbolUniform
adata = sc.read('Data/liver.h5ad', backup_url='https://cloud.tsinghua.edu.cn/f/45a7fd2a27e543539f59/?dl=1')
adata

2.加用统一的基因(42117个基因)转化原始的 h5ad
这一步将输入adata中的基因转换为42117个基因符号,并保留相应的基因表达值。
adata_GS_uniformed = GeneSymbolUniform(input_adata=adata,
output_dir="Data/",
output_prefix='liver')
3.处理统一数据(简单的norm和log1p)
adata_GS_uniformed
# norm and log1p count matrix
if adata_GS_uniformed.X.max() > 10:
sc.pp.normalize_total(adata_GS_uniformed, target_sum=1e4)
sc.pp.log1p(adata_GS_uniformed)
4.载入scMulan模型
ckp_path = 'ckpt/ckpt_scMulan.pt'
scml = scMulan.model_inference(ckp_path, adata_GS_uniformed)
base_process = scml.cuda_count()
scml.get_cell_types_and_embds_for_adata(parallel=True, n_process = base_process)
5.可视化
adata_mulan = scml.adata.copy()
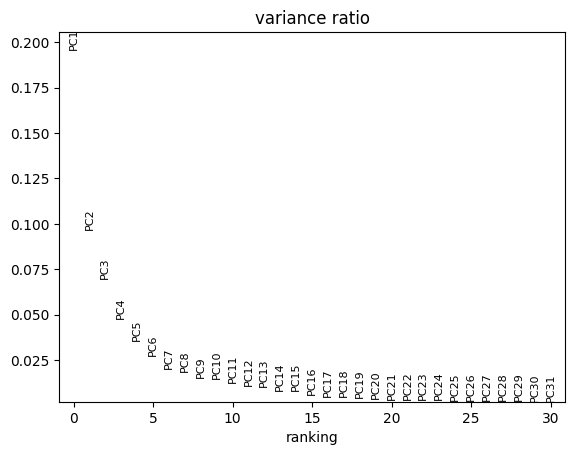
sc.pp.pca(adata_mulan)
sc.pl.pca_variance_ratio(adata_mulan)
sc.pp.neighbors(adata_mulan,n_pcs=10)
sc.tl.umap(adata_mulan)
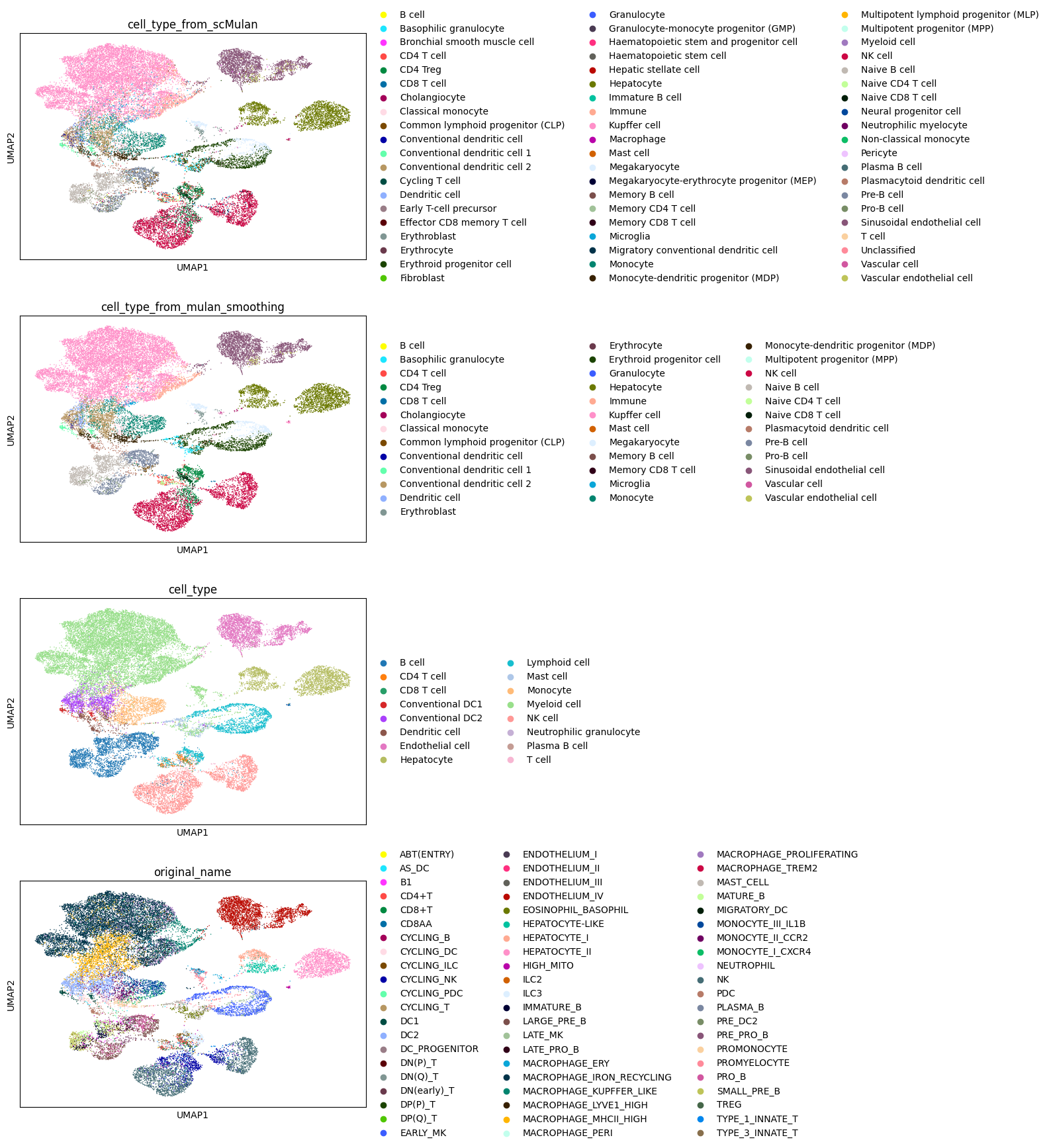
scMulan.cell_type_smoothing(adata_mulan, threshold=0.1)
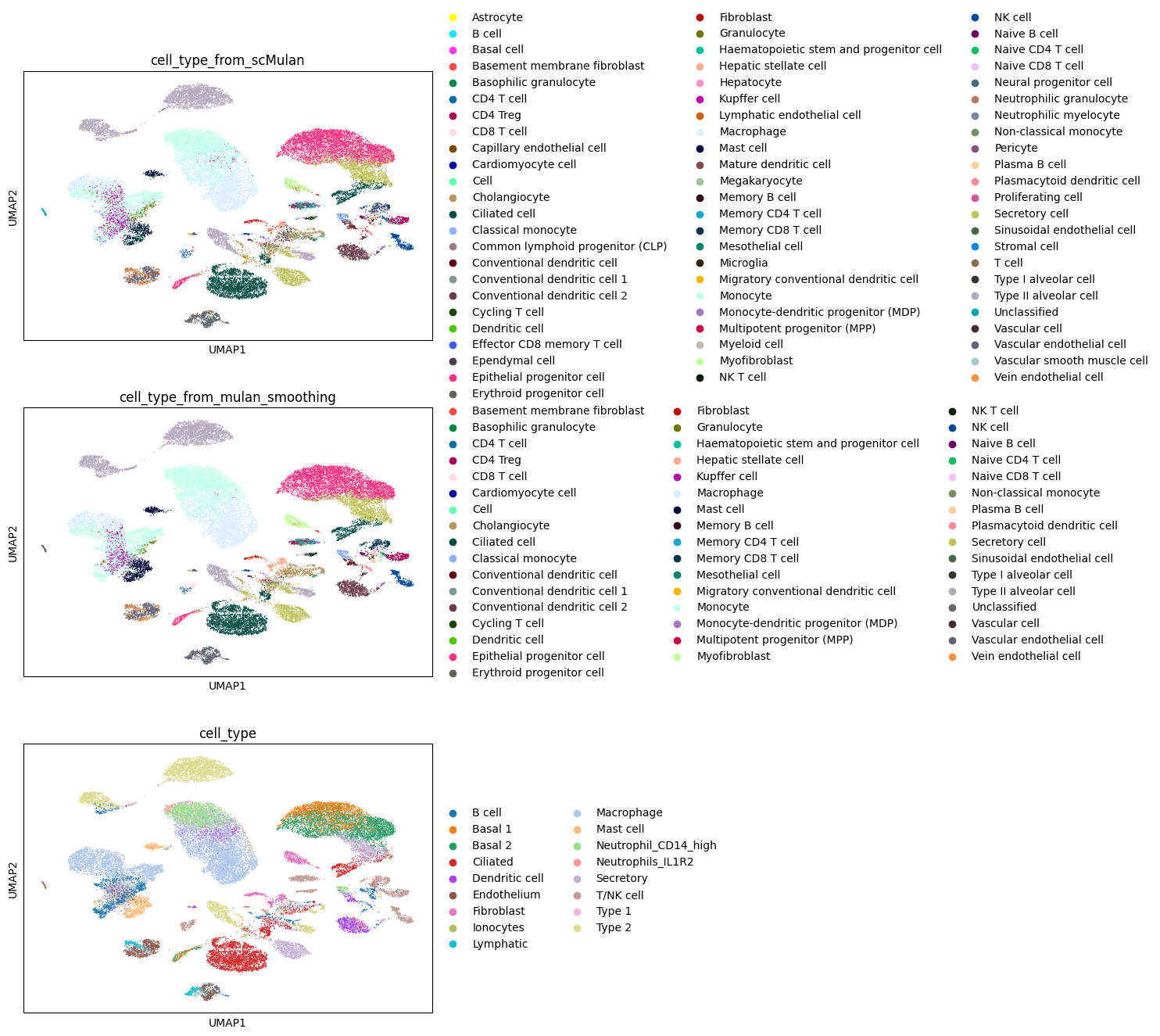
sc.pl.umap(adata_mulan,color=["cell_type_from_scMulan","cell_type_from_mulan_smoothing",'cell_type','original_name'],ncols=1)
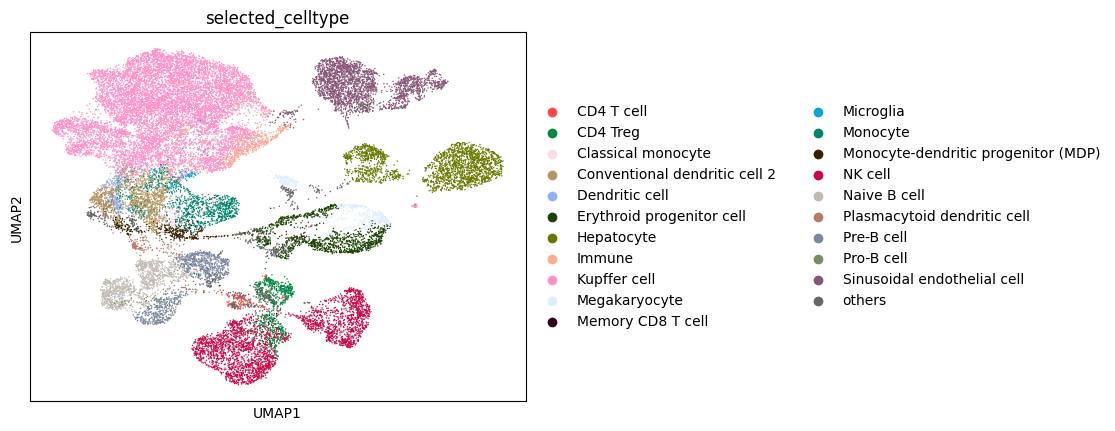
top_celltypes = adata_mulan.obs.cell_type_from_scMulan.value_counts().index[:20]
selected_cell_types = top_celltypes
scMulan.visualize_selected_cell_types(adata_mulan,selected_cell_types,smoothing=True)

3.2 zeroshot 数据整合
代码来源:https://github.com/SuperBianC/scMulan/blob/main/Tutorial-integration.ipynb
数据提供(肺):https://figshare.com/ndownloader/files/24539942
1.导入数据
import os
os.environ["CUDA_VISIBLE_DEVICES"] = "0,1,2,3" ## 每个GPU用约2G的运行内存
import scanpy as sc
adata = sc.read(
"Data/lung_atlas.h5ad",
backup_url="https://figshare.com/ndownloader/files/24539942",
)
adata
2.用统一的基因(42117个基因)转化原始 h5ad
adata_GS_uniformed = GeneSymbolUniform(input_adata=adata,
output_dir="Data/",
output_prefix='lung')
3.处理统一数据(简单的norm和log1p)
adata_GS_uniformed
# norm and log1p, if adata is count matrix
if adata_GS_uniformed.X.max() > 10:
sc.pp.normalize_total(adata_GS_uniformed, target_sum=1e4)
sc.pp.log1p(adata_GS_uniformed)
4.载入模型
# https://cloud.tsinghua.edu.cn/f/2250c5df51034b2e9a85/?dl=1
ckp_path = 'ckpt/ckpt_scMulan.pt'
scml = scMulan.model_inference(ckp_path, adata_GS_uniformed)
base_process = scml.cuda_count()
scml.get_cell_types_and_embds_for_adata(parallel=True, n_process = base_process)
5.可视化
adata_embds = sc.AnnData(scml.adata.obsm['X_scMulan'].copy())
adata_embds.obs = scml.adata.obs
adata_embds
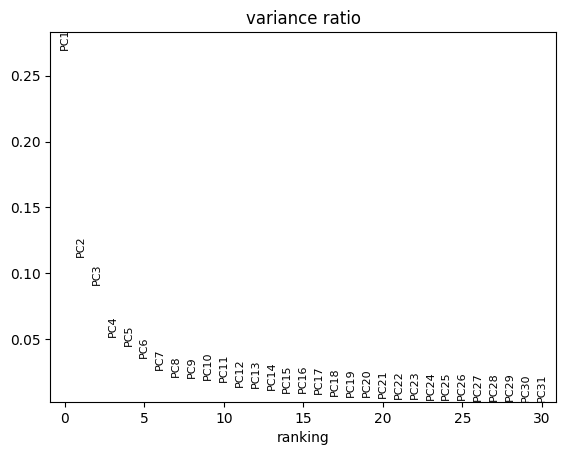
sc.pp.pca(adata_embds)
sc.pl.pca_variance_ratio(adata_embds)
sc.pp.neighbors(adata_embds,n_pcs=12)
sc.tl.umap(adata_embds)
scMulan.cell_type_smoothing(adata_embds, threshold=0.1)
sc.pl.umap(adata_embds,color=["cell_type_from_scMulan","cell_type_from_mulan_smoothing",'cell_type'],ncols=1)
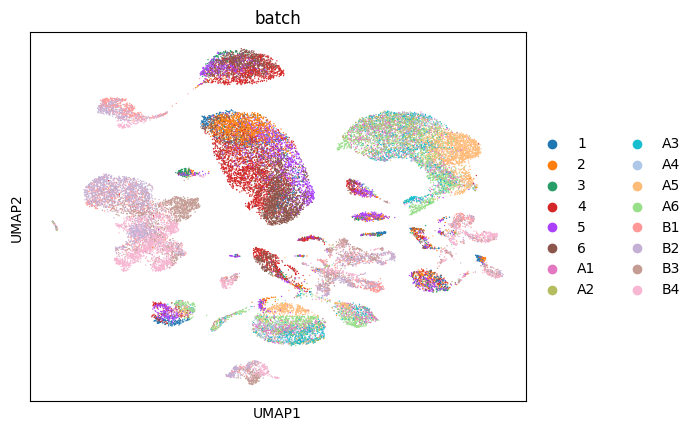
sc.pl.umap(adata_embds, color = ['batch'])




















 1262
1262

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











