






 该博客介绍了如何利用GMAP代替blastn来构建ALLHiC所需的Allele.ctg.table。首先,需要准备多倍体基因组的草图序列和近缘物种的cds序列。然后,通过gmap_build命令创建GMAP索引,接着使用GMAP进行比对生成gff3文件。最后,利用gmap2AlleleTable.pl脚本来生成allelic.ctg.table。这种方法无需基因组注释,简化了流程。
该博客介绍了如何利用GMAP代替blastn来构建ALLHiC所需的Allele.ctg.table。首先,需要准备多倍体基因组的草图序列和近缘物种的cds序列。然后,通过gmap_build命令创建GMAP索引,接着使用GMAP进行比对生成gff3文件。最后,利用gmap2AlleleTable.pl脚本来生成allelic.ctg.table。这种方法无需基因组注释,简化了流程。
在ALLHiC使用过程中需要构建Allele.ctg.table,用于过滤多倍体基因组中因等位序列相似引起的HiC噪音的必要输入。官网提供了两种办法,一种是blastn,需要对草图基因组进行注释,这个过程挺麻烦的,在最下边看到了也可以使用GMAP。我的目录下之前已经安装了GMAP, 可我对这个软件已经一点印象也没有了,再学习一下。
GMAP的方法不需要提供目标基因组的注释文件,只需要提供多倍体基因组的基因组序列和近缘物种的cds序列即可,github上的方法介绍链接:
https://github.com/tangerzhang/ALLHiC/issues/16
一.GMAP简介
GMAP是一款比对软件,与bowie和bwa类似,能够将DNA片段mapping到基因组上的软件,最早用于将EST/cDNA序列比对到参考基因组上,可以用于基因组结构注释。后来又开发了GSNAP支持高通量数据比对。PacBio测序技术出现后,GMAP常用于Iso-Seq全长转录本的比对。
二.GMAP构建Allele.ctg.table
1.准备近源物种的cds序列和自己的草图基因组序列
2.GMAP对草图基因组建立索引
gmap_build -D path -d dbname draft.genome.fasta
参数说明:
-D 创建索引的存放路径(默认存放在安装路径下的share文件夹);
-d 创建索引的名字;。
3.GMAP生成gff3文件
map -D path -d dbname -t 12 -f 2 -n $N reference.cds.fasta > gmap.gff3
参数说明:
-t 表示使用多少条线程进行计算,默认是1;
-D 参考序列索引的位置;
-d 参考序列索引的名字;
-n 草图基因组来源物种的染色体倍性;
-f 输出格式,输出的gff3格式,-f 有1-9个选择;
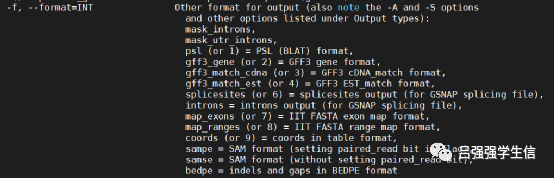
4. 生成allelic.ctg.table
gmap2AlleleTable.pl gmap.gff3
注意:*.gff3文件前缀一定要是gmap,要不然这个Perl脚本会不认识gff3文件。
5.结果allelic.ctg.table
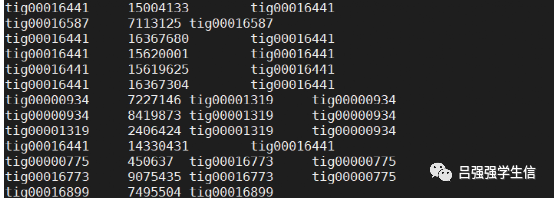
参考:
ALLHiC续: 如何构建Allele.ctg.table_徐洲更hoptop的博客-CSDN博客
https://blog.csdn.net/u012110870/article/details/102943821

















 5630
5630

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









