







 本文介绍了单细胞转录组分析中的scVI技术,一种基于神经网络和贝叶斯模型的单细胞变分推理方法,用于处理噪声、非正态分布和批次效应。scANVI则进一步扩展了这一框架,通过半监督学习整合和注解单细胞数据。文章强调了这些技术在生物学研究中的作用以及它们在处理大规模单细胞数据集时的可扩展性。
本文介绍了单细胞转录组分析中的scVI技术,一种基于神经网络和贝叶斯模型的单细胞变分推理方法,用于处理噪声、非正态分布和批次效应。scANVI则进一步扩展了这一框架,通过半监督学习整合和注解单细胞数据。文章强调了这些技术在生物学研究中的作用以及它们在处理大规模单细胞数据集时的可扩展性。
文章目录
来自Manolis Kellis教授(MIT计算生物学主任)的课
YouTube:Single Cell Genomics - Lecture 10 - Deep Learning in Life Sciences (Spring 2021)
Slides: slides
本节课是三个部分,这篇是第三部分。
本部分是邀请UC Berkeley的Lopez来介绍单细胞组学领域相关的工作。跟第二部分比较类似。主要的工作的介绍基本涵盖了这方面的分析流程。


8.1 Background & Review
8.1.1 Single-cell Transcriptomics
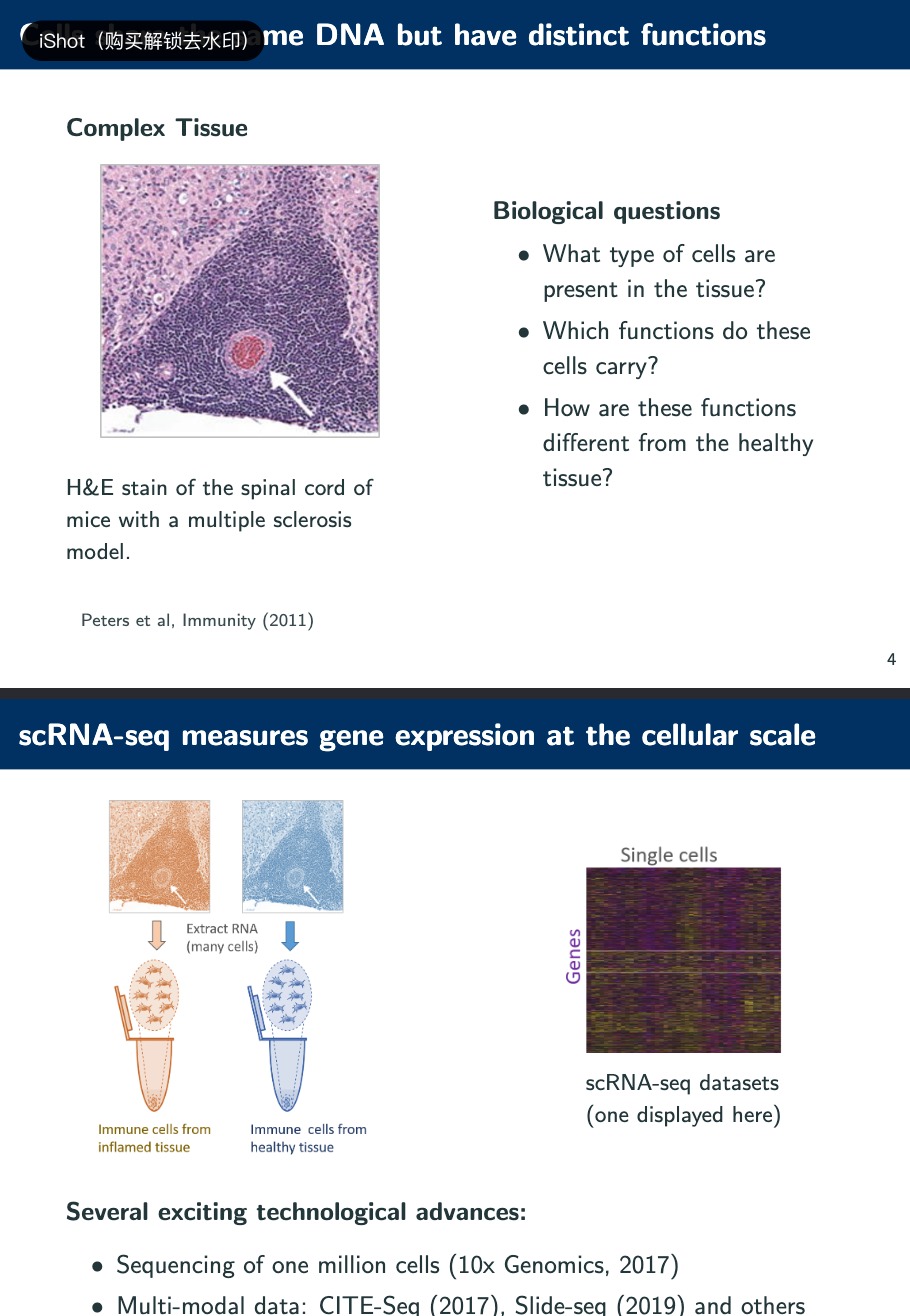
对于一个疾病(多发性硬化症模型),有正常的组织和坏掉/发炎的组织,如何研究这个组织从正常到发病的过程,就需要scRNA-seq来为我们揭示
分别从发炎组织和健康组织中提取免疫细胞,并获得单个细胞的基因表达模式

扰动/蛋白质/转录组
大部分生物学问题可以转化为cell-level或gene-cell-level水平的算法查询
列出了几个计算生物学任务及其定义:
-
cell-level
-
分层via嵌入(Stratification via Embedding):将细胞投影以进行识别,如聚类或轨迹分析等。
-
和谐化(Harmonization):提供一个无批次效应的embedding,以比较不同条件下的数据。
-
注释(Annotation):将一个数据集中的细胞类型标签转移到另一个数据集。
-
-
Gene-cell level
- 标准化/填充(Normalization/Imputation):计算平均表达水平,同时去除技术性偏差。
- 差异表达(Differential Expression):找到不同细胞类型之间的基因表达差异。

一些挑战
- 噪声
- 可变的测序深度
- 批次效应
- scRNA-seq数据(每个细胞中每个基因的RNA分子数)数据不符合正态分布,能直接应用假设正态分布的传统统计方法
- 可扩展性:未来能测序的数据越来越多

pipeline:数据标准化(比如让其符合高斯分布)->降维->校正批次效应->聚类->差异表达分析
提出一个问题:“如何在整个数据处理流程中找到统一的建模假设?”
在scRNA-seq数据分析中,由于数据的复杂性和高维度,这是一个重要但困难的任务。

这里说明了为什么PCA不完全适用于单细胞RNA测序(scRNA-seq)的分析
- PCA的数据假设

问题
- scRNA-seq数据通常是非正态分布的,而且具有偏斜性质
- 基因表达与潜在生物学过程之间的关系可能是非线性的,而传统PCA基于线性关系的假设
- 传统PCA假设噪音很小,实际scRNA-seq数据的噪音很大

- 可扩展和一致的框架,用于全概率分析scRNA-seq数据
- scVI将生成一个适合scRNA-seq数据分布的模型
scVI能够通过利用随机优化来轻松解决所有任务并且易于扩展。随机优化是一种考虑优化过程中随机性(或称为随机性)的优化技术,适用于在存在不确定性时寻找最优解。在scRNA-seq数据分析中,这种方法特别有用,因为数据通常包含了大量的生物学和技术变异性。
8.1.2 Bayesian Modeling
在李宏毅的VAE教程里我已经学过了,就不记了这里
这里涉及变分推理,用于估计概率模型中的概率分布。特别是当模型复杂到无法直接计算后验分布。
在贝叶斯统计中,我们通常对隐变量或模型参数的后验分布感兴趣。理论上,我们可以通过贝叶斯定理直接计算这个后验分布。然而,对于复杂的模型,这个分布可能是无法解析计算的,因为它涉及到高维积分,这在数学上称为不可解析或不可逾越。
变分推理通过提出一个简单的分布来近似后验分布来解决这个问题。这个简单的分布被选为容易计算的分布,通常是因子分解的(意味着它由更小的分布的乘积组成),并且有足够的灵活性来近似真实的后验分布。
变分推理的目标是找到这个简化分布的参数,使其尽可能接近真实的后验分布。这是通过优化一个称为变分下界(或证据下界,ELBO)的目标函数来实现的。ELBO是模型证据的一个下界,它量化了变分分布和真实后验分布之间的相似度。通过最大化ELBO,我们可以改善变分分布,使其更接近真实的后验分布。
8.2 Single-cell Variational Inference(scVI)
模型用神经网络来处理这些数据,目的是要尽量去除技术效应的干扰,从而得到更准确的生物学信号。一旦得到了准确的信号,就可以用来做很多不同的生物学分析,比如找出哪些基因的活跃程度在不同的条件下会变化。
- 使用统计建模以下数据
- cell embedding
- 文库大小:基因测序时用到的一种技术校正方法
- 标准化基因表达数据
- Raw data
这里用神经网络来整合先验分布(如 Z n Z_n Zn、 l n l_n ln)和实验数据(如 X n g X_{ng} Xng、 S n S_n Sn),通过训练数据来估计 $ρ_n $的后验分布

通过使用统一的模型,所有这些任务都基于相同的数据表示和假设,这有助于避免在使用不同模型进行分析时可能出现的不一致性和统计伪迹。
在贝叶斯统计中,这些任务中的一些涉及后验抽样和条件化:
- 后验抽样是指在观察到数据之后,对模型参数(如 Z n Z_n Zn 和 S n S_n Sn)的分布进行抽样,以了解其可能的取值范围和不确定性。
- 条件化是指在给定某些信息(如批次信息 S n S_n Sn或隐藏标量 l n l_n ln)的情况下,分析其他数据或参数。
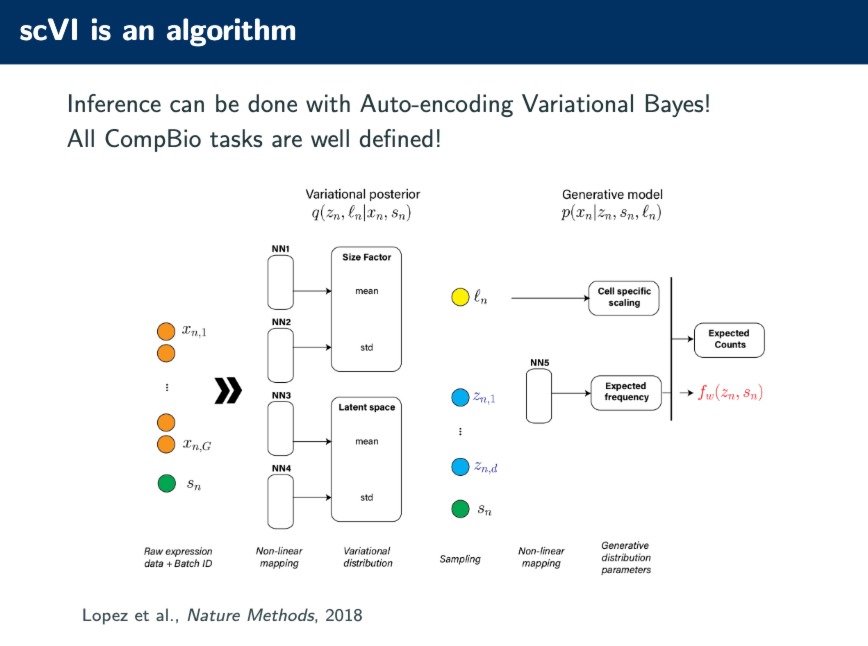
scVI用于推断单细胞基因表达数据。
算法由两部分组成:变分后验(Variational posterior)和生成模型(Generative model)。
-
变分后验:
- 这部分的目的是估计隐变量 ( Z n Z_{n} Zn ) 和 ( $\ell_{n} KaTeX parse error: Can't use function '\)' in math mode at position 1: \̲)̲ 的后验分布,也就是在给定观察… X_{n,G}$ ) 和批次信息 ( S n S_n Sn ) 后,隐变量的条件分布。
- 这里使用了神经网络(NN1到NN4)来近似这个分布。NN1用于计算文库大小(Size Factor)的均值和标准差,NN3和NN4用于计算潜在空间(Latent space)的均值和标准差,这是细胞状态的压缩表示。
- 通过这些网络,模型可以将原始的表达数据和批次信息映射到一个更易于处理的形式,从而可以更好地进行采样和后续的分析。
-
生成模型:
- 这部分使用隐变量来生成(或者说重构)观测数据的分布。
- 神经网络NN5用于根据潜在空间 ( Z n Z_{n} Zn ) 和批次信息 ( S n S_n Sn ) 的参数来预测细胞特异性的表达频率(Expected frequency)。
- 预测的频率和文库大小 ($ \ell_{n}$ ) 结合使用,来估计基因表达的预期计数( E x p e c t e d C o u n t s Expected Counts ExpectedCounts)。
scVI算法通过对单细胞RNA序列数据中的隐变量进行建模,并使用VAE的框架来进行推断,从而能够从数据中学习到有用的表示,并用于多种下游分析任务
能够处理单细胞数据中的技术噪声,并且能够适应不同的实验条件和批次效应。
Z n Z_n Zn
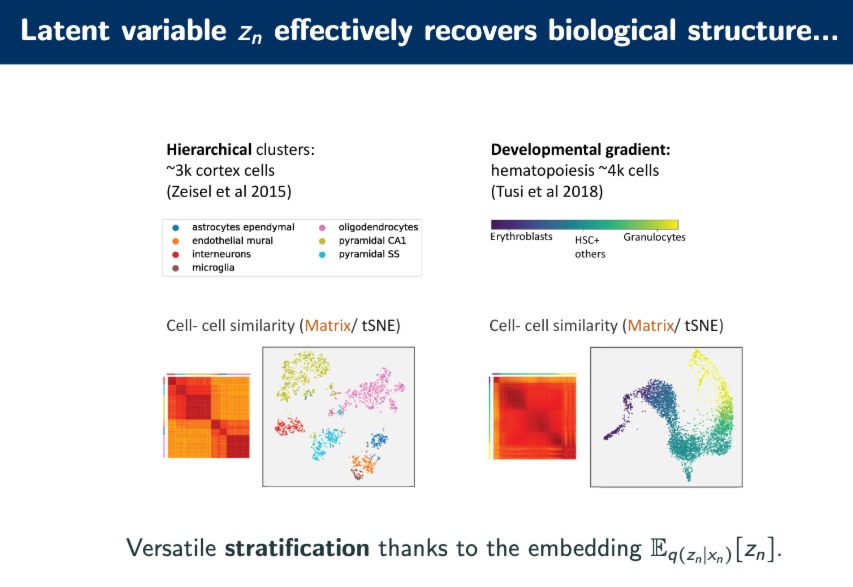
Zn是embedding,中间的低维表示,基于Zn进行了分类
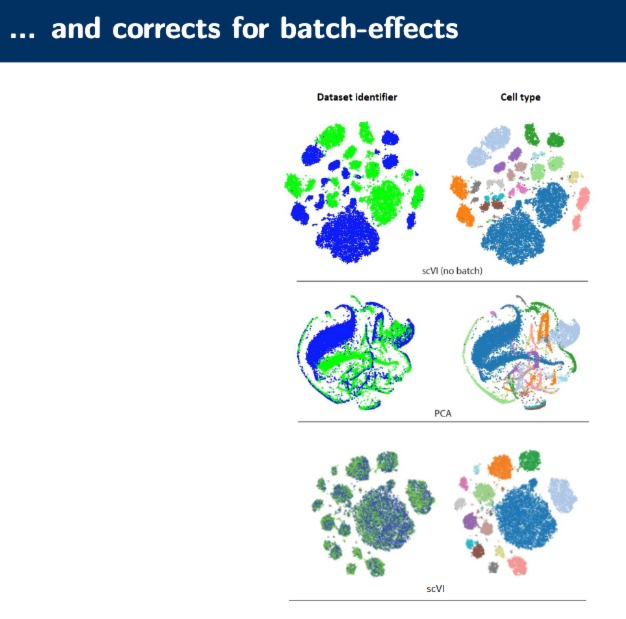
scVI可以较好的解决批次效应带来的问题
ρ n ρ_n ρn
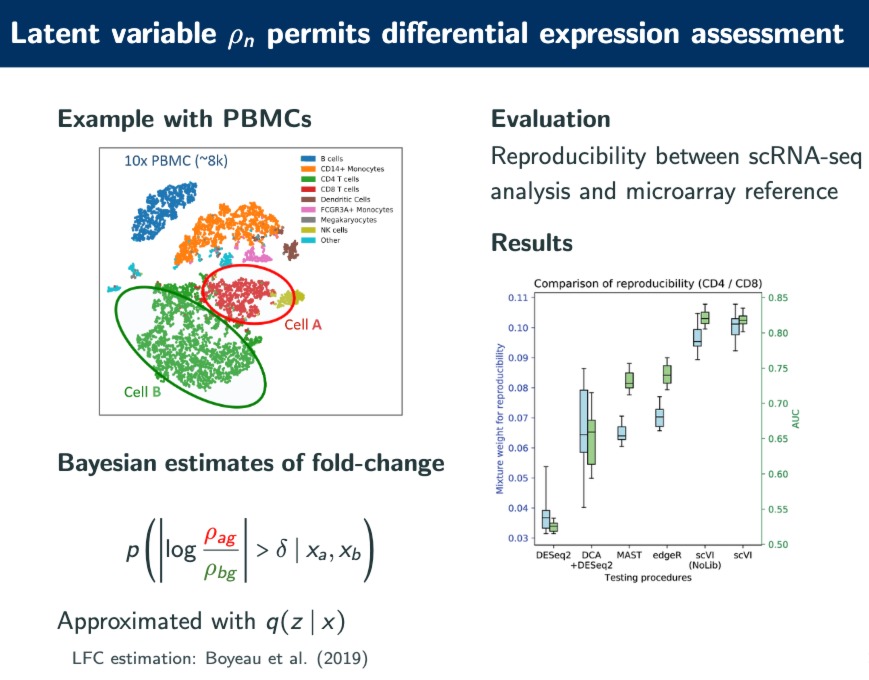
用于差异表达分析
对圈出来的cell A和B,进行差异表达分析。右边展示了不同的差异分析方法的对比
公式表示用Bayesian估计基因表达变化的概率。
具体来说,它表示了在给定两组细胞( x a x_a xa 和 x b x_b xb的条件下,观察到基因 ( g ) 的表达变化超过某个阈值 ( δ \delta δ ) 的概率。
这里的 $ p $ 表示概率密度函数,而 $q(z|x) 表示给定观测数据 表示给定观测数据 表示给定观测数据 x $ 时潜在变量 z z z 的后验分布,这通常由VAE(如scVI)提供的近似值。LFC(Log Fold Change)估计是用来测量两个条件(例如不同细胞类型)之间基因表达水平的相对变化。
- 计算折叠变化(Fold Change):使用VAE的潜在变量来计算两个条件下基因表达的折叠变化(Fold Change),这可以是对数折叠变化(Log Fold Change)。
- Bayesian估计:VAE的Bayesian框架可以用来估计差异表达的不确定性,提供比传统方法更为丰富的统计推断
scVI在单细胞数据分析中的应用,特别是在识别和量化基因表达变化方面
可扩展性
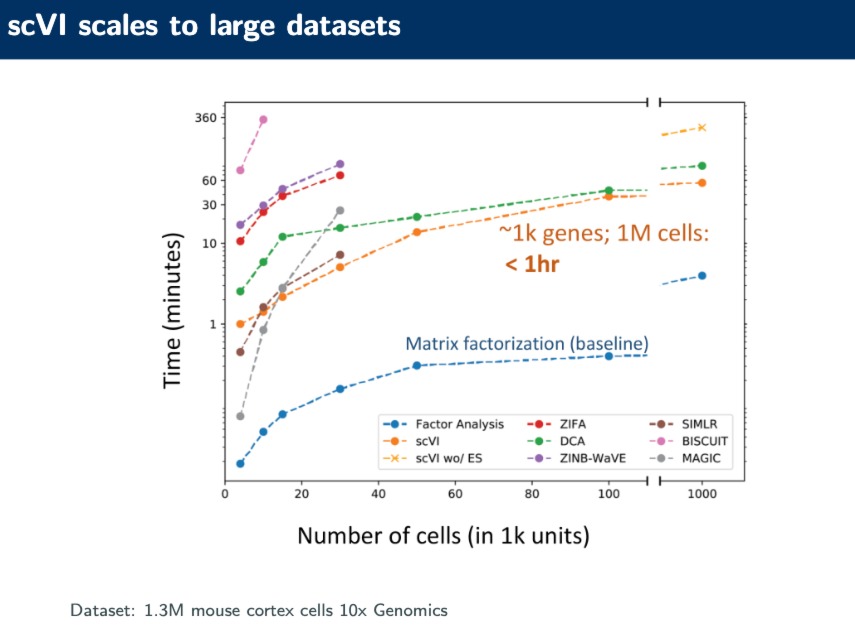
8.3 Probabilistic annotation(scANVI)

scANVI旨在提供一个统一的框架来处理单细胞数据的整合和注释,可能是通过学习数据的生成模型来实现的,并且可以在没有额外信息的情况下估计差异表达。
将不同批次、实验室、技术的数据混合到一起
半监督学习,有些有标签,有些没有。
在这里是对于存在部分注释的数据集A,把全部注释的数据集B的注释,传递给A。

scANVI的方法:
- 改变潜在变量 z z z的先验分布,从各向同性正态分布变为混合模型。这意味着潜在空间不再假设所有维度上的变异是相同的,而是可以根据混合模型来捕捉更复杂的数据结构。
- 将半监督学习看作是混合分配(例如,细胞类型 c c c)的缺失值问题。这使得算法能够在部分注释的数据集中推断出未标记细胞的类型。
scANVI作为一个概率性的端到端方法,利用半监督学习来处理单细胞数据的注释问题,尤其是在整合来自不同批次或实验室的数据时。这种方法可以减少人工注释的需求,提高注释过程的效率和准确性。
https://scvi-tools.org
他们的一篇综述:[Enhancing scientific discoveries in molecular biology with deep generative models](


















 874
874

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











