







 本文介绍了动态互相关矩阵(DCCM)的重要性和计算过程,包括数学概念、常用工具如Bio3D和GROMACS的gmx covar命令。详细解释了DCCM的计算步骤,涉及残基协方差、相关性系数,以及如何通过gmx covar绘制DCCM图。此外,还提到了其他DCCM计算工具如MD-TASK和Correlationplus,并展示了代码复现DCCM计算的部分细节。
本文介绍了动态互相关矩阵(DCCM)的重要性和计算过程,包括数学概念、常用工具如Bio3D和GROMACS的gmx covar命令。详细解释了DCCM的计算步骤,涉及残基协方差、相关性系数,以及如何通过gmx covar绘制DCCM图。此外,还提到了其他DCCM计算工具如MD-TASK和Correlationplus,并展示了代码复现DCCM计算的部分细节。
关注公众号【杜艾维】免费获取本文~
0. Preface
对于关键残基的识别、配体结合、突变效应和长距离相互作用等过程,从大分子动力学轨迹中提取的动态互相关性信息是非常重要的。
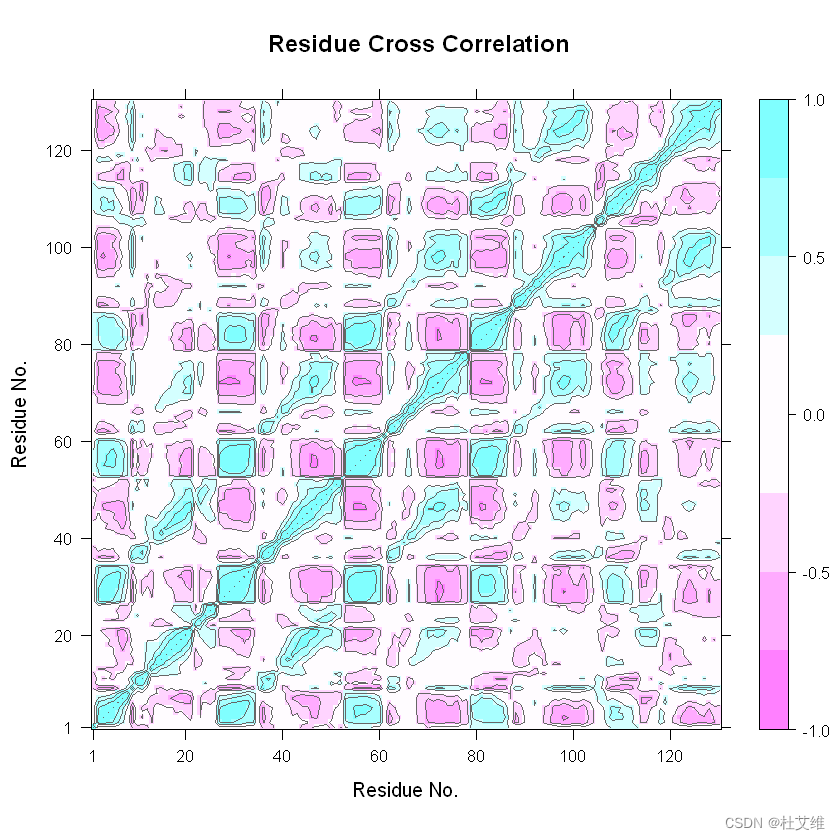
下图即是一张Bio3D绘制的某蛋白的动态互相关矩阵(DCCM)图。
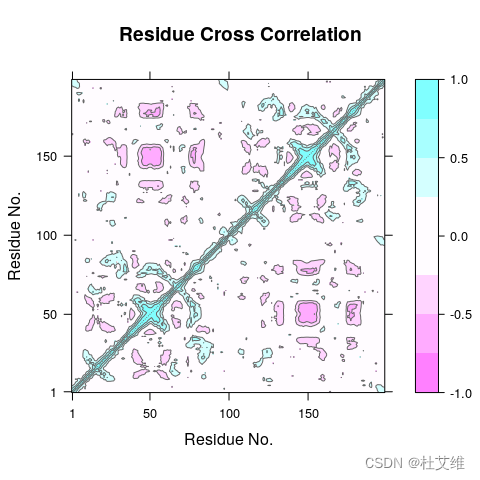
做蛋白质相关的模拟,难免经常遇到有需要分析DCCM的场合,这篇小文就简单介绍DCCM相关的一点儿内容,包括数学过程以及目前能够计算DCCM的几种工具。
1. 一些儿数学过程
我们都很清楚方差的概念:
σ ( x , x ) = < ( x − x ˉ ) 2 > \sigma (x, x) = < (x - \bar x)^2 > σ(x,x)=<(x−xˉ)2>
也即我们有一个变量 x x x,其可以取很多值。 x x x的方差就定义为每一个取值与平均值的差的平方,最后再求平均(尖括号表示平均)。
协方差的概念也是类似的,比如我们有两个变量 x x x和 y y y:
c ( x , y ) = < ( x − x ˉ ) ⋅ ( y − y ˉ ) > c(x, y) = < (x - \bar x)·(y - \bar y)> c(x,y)=<(x−xˉ)⋅(y−yˉ)>
方差衡量的是变量的离散程度,协方差衡量的是两个变量的联合变化程度;也即如果两个向量表现出相似的行为,则协方差为正。
协方差虽然能够表征两个变量的联合变化,但是其绝对数值(变量的变化幅度)会影响我们对于相关性的观察。因而还有相关性系数的计算:
C ( x , y ) = c ( x , y ) ( σ x σ y ) 1 / 2 C(x, y) = \frac{c(x, y)}{(\sigma_x \sigma_y)^{1/2}} C(x,y)=(σxσy)1/2c(x,y)
相关性系数即是剔除了两个变量变化幅度的影响,标准化处理之后的协方差。其反映的就是纯粹的两个变量的相关性了。
动态互相关矩阵(dynamical cross-correlation matrix, DCCM)涉及到的数学过程尚属简易的,基本就是上面这些了。
我们想要获知成对残基之间的运动模式关联,可以计算成对残基之间的协方差。
公式如下:
c ( i , j ) = < Δ R i ⋅ Δ R j > c(i,j) = <\Delta R_{i} · \Delta R_{j} > c(i,j)=<ΔRi⋅ΔRj>
其中: Δ R i = R i − < R i > \Delta R_{i} = R_{i} - < R_{i} > ΔRi=Ri−<Ri>
i i i和 j j j表示的是蛋白质氨基酸残基的序号,也即这里的 c ( i , j ) c(i, j) c(i,j)是残基 i i i和残基 j j j的协方差。尖括号表示的是系综平均,也即计算的是一系列模型的平均,包括模拟轨迹里面的多帧构象、NMR得到的多个结构等等。 Δ R i \Delta R_{i} ΔRi表示的是位置偏移量,且是基于系综平均位置的偏移量。
所谓的动态互相关矩阵,也就是把残基对之间的协方差转换为互相关指数,并按残基编号组织起来,成为一个矩阵而已。
C ( i , j ) = c ( i , j ) [ c ( i , i ) ⋅ c ( j , j ) ] 1 / 2 C(i, j) = \frac{c(i, j)}{[c(i, i)·c(j, j)]^{1/2}} C(i,j)=[c(i,i)⋅c(j,j)]1/2c(i,j)
当然,就像我们在PCA之前要进行平动和转动的去除一样,在计算DCCM之前也需要对齐结构,避免体系整体的运动掩蔽掉体系内部的运动。
最后,再简单阐述下对一段儿常规模拟轨迹计算DCCM的过程吧。
例如我们的蛋白质有N个残基,要计算残基alpha-C原子的DCCM。我们首先要做的是对这段儿轨迹进行周期性校正,去除平动转动。之后需要将每一帧的原子坐标对齐到参考结构,得到对齐之后的每一帧的原子坐标。然后对于每一个C原子,计算其在这段轨迹中的平均位置。有了这个平均坐标,就可以计算每一帧中这个C原子相对于其平均位置的偏移向量(x, y, z)了。之后对于每一帧轨迹,计算两两C原子偏移量之间的点积,也即协方差,然后再将多帧的协方差对帧数取平均。如此我们就得到两两残基之间在这段模拟轨迹中的协方差矩阵了。最后,对协方差矩阵上的元素计算相关性系数就可以得到动态互相关矩阵。
2. 于浩然老师的教程
关于如何利用Bio3D计算DCCM,不仅Bio3D官方教程可以作为参考,还可以参考于浩然老师20年发表的文章A beginner’s guide to molecular dynamics simulations and the identification of cross-correlation networks for enzyme engineering。这篇文章从一个具体的蛋白实例开始,阐述模拟构建、分析以及利用Bio3D绘制DCCM的过程。细节丰富详实,容易上手,并且文中有大量的关于DCCM含义、用途和意义的阐述,很值得一看。当然也可以辅以于老师18年的PNAS文章Coupled molecular dynamics mediate long- and short-range epistasis between mutations that affect stability and aggregation kinetics,在实际案例中体会DCCM的作用。
3. 利用gmx covar命令绘制DCCM
在于老师20年的文章中也提到,除了Bio3D,GROMACS本身的命令covar也可以计算得到协方差矩阵,但是还需要一点儿处理才能获得动态互相关矩阵。
covar命令用于计算并对角化(质量加权)的协方差矩阵,所有结构都会叠合到结构文件中的参考结构。计算得到的本征向量会写到轨迹文件中,这我们在之前的PCA与FEL绘制一文中已经遇到过。而计算得到的协方差矩阵会通过-xpm和-xpma写到xpm文件中。-xpma得到的xpm文件包含了每个原子对三个坐标的协方差的总和,因而是
N
∗
N
N*N
N∗N的,N为原子数目。而-xpm得到的xpm则是包含每一维坐标的,因而是
3
N
∗
3
N
3N*3N
3N∗3N的。要做后处理,当然或许可以直接读取xpm文件的数据然后计算互相关指数得到DCCM;也可以通过covar命令的-ascii参数输出整个的协方差矩阵到一个文本文件(
3
N
∗
3
N
3N*3N
3N∗3N),如此得到的数据或许会稍微精准一些。之后就只需要读取这个文本文件的数据,处理一下就可以了。有幸,有现成的代码可以用!https://github.com/busrasavas/gmx_corr
首先我们需要利用covar命令得到协方差矩阵:
gmx covar -s md.tpr -f md_fit.xtc -o eigenvalues.xvg -v eigenvectors.trr -xpma results.xpm -ascii results.dat
按照需要选择对齐的组和计算的组,之后协方差矩阵会被保存到xpm文件中,协方差矩阵的数据信息会被保存到dat文件中。
得到的协方差矩阵可视化之后:
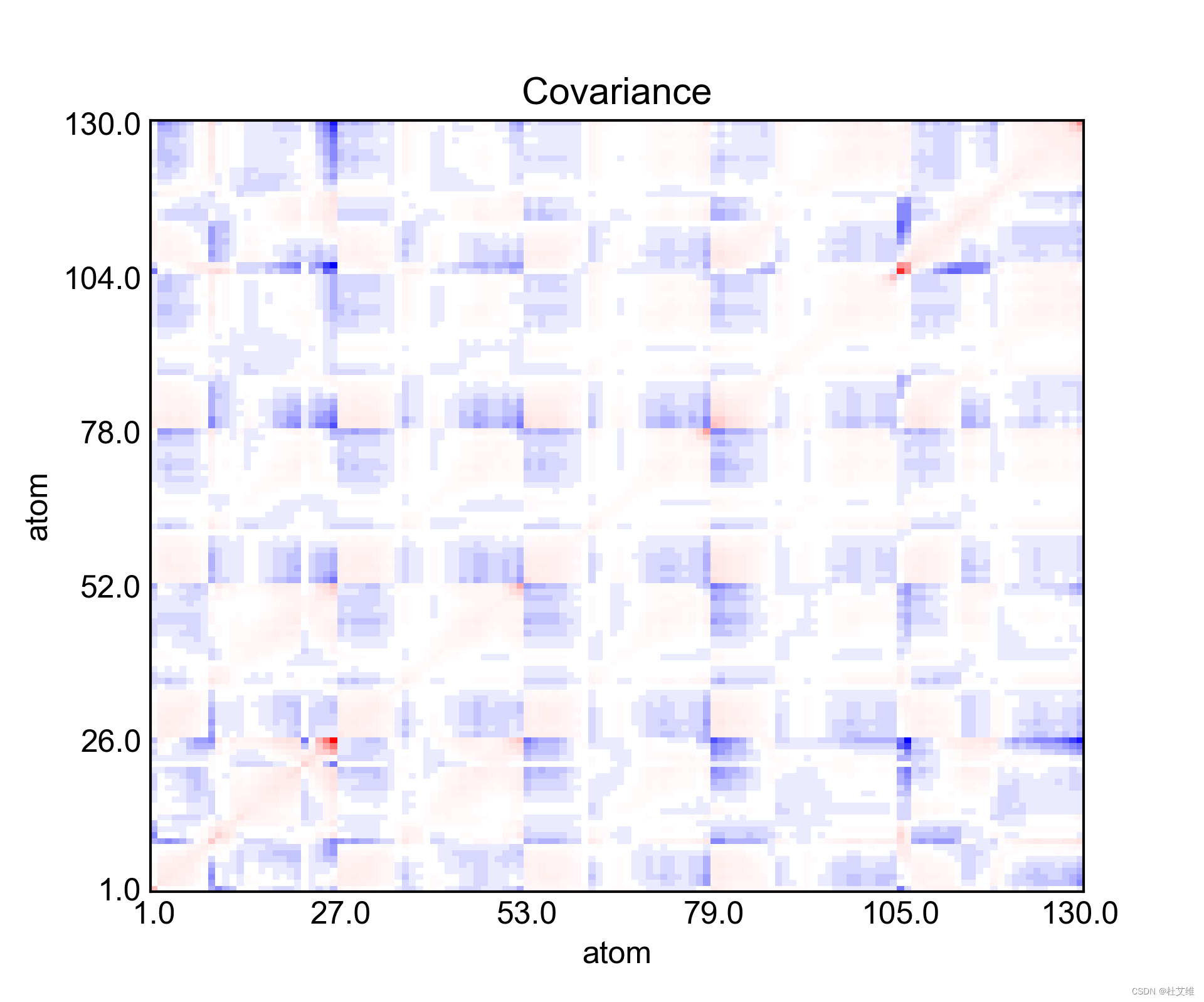
如前所言,协方差矩阵不仅包含了相关性信息,还有波动性信息在里面。其中左下到右上的对角线的点,表示的就是当前氨基酸的波动性信息,越红,波动性越强。
covar命令保存协方差矩阵数据到dat文件的方式有些奇怪,我以一个具体例子进行说明吧。
假设2个原子计算得到的协方差矩阵如下(6*6):
1 2 3 4 5 6
7 8 9 10 11 12
13 14 15 16 17 18
19 20 21 22 23 24
25 26 27 28 29 30
31 32 33 34 35 36
-xpm参数输出的矩阵就像这个样子,但是在输出到dat文件中的时候,每一行只保留了三个数字,顺序是从左到右、从上到下的输出这个矩阵,会得到:
1 2 3
4 5 6
7 8 9
10 11 12
13 14 15
16 17 18
19 20 21
22 23 24
25 26 27
28 29 30
31 32 33
34 35 36
只要知道dat文件里面数据的组织方式,就很好进行处理了。
3 N ∗ 3 N 3N*3N 3N∗3N的矩阵如何处理成 N ∗ N N*N N∗N的矩阵呢?很简单,对每一个 3 ∗ 3 3*3 3∗3的矩阵求这9个数的总值就行了。
原理如此,执行计算的代码之后也会分享给大家。最后将矩阵可视化就可以得到最后的动态互相关图了。
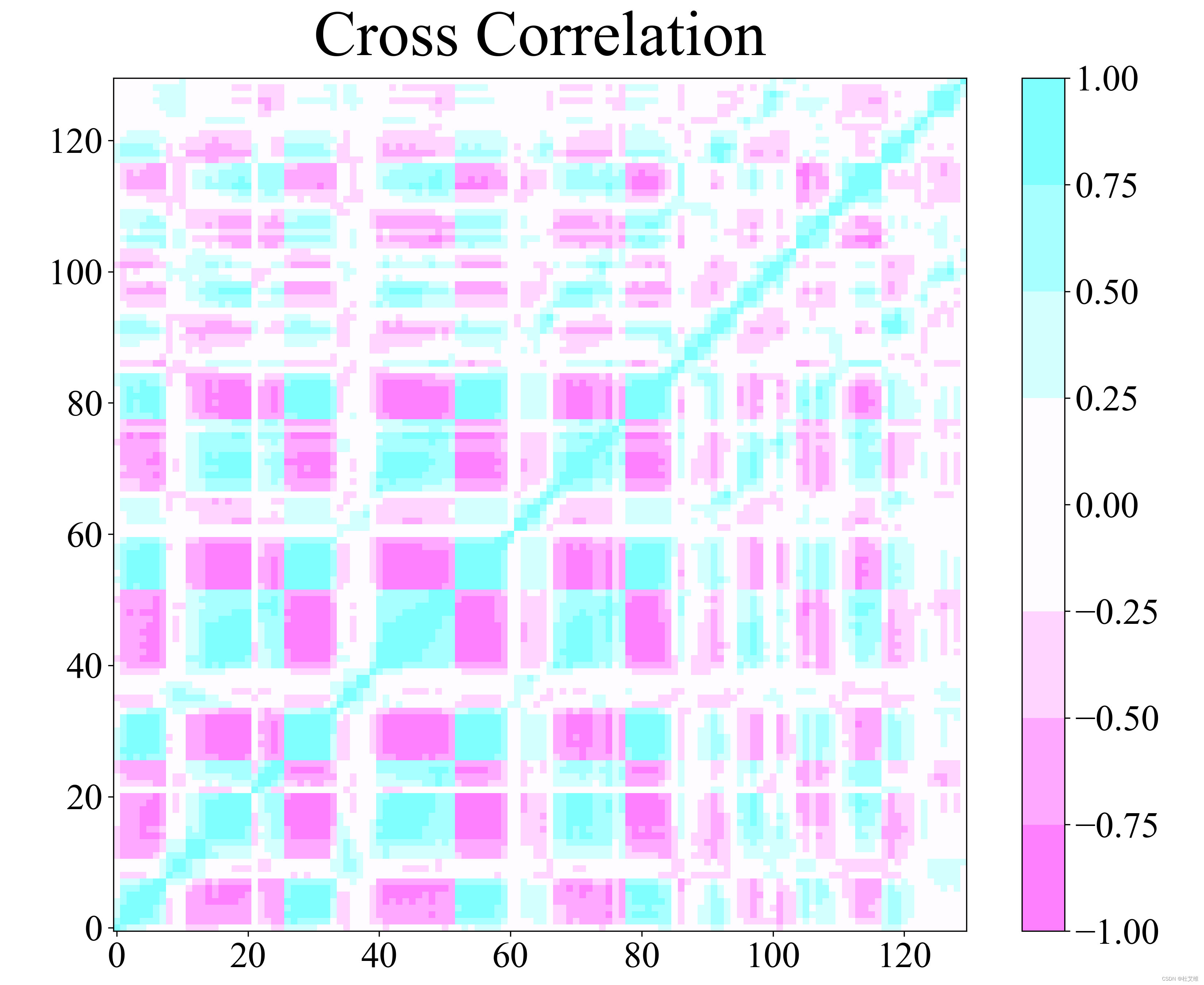
本征向量绘制豪猪图
这部分和DCCM关系不大,但是值得一提。我们可以利用anaeig命令从协方差矩阵中导出特征向量。这些特征向量可以用于绘制自由能形貌图,也可以通过绘制豪猪图去直观地观察轨迹沿主成分方向的投影。
比如说我们导出第一个主成分并写入到pdb文件中:
gmx anaeig -s md.tpr -f md.xtc -v eigenvectors.trr -eig eigenvalues.xvg -proj proj_ev.xvg -extr ev.pdb -rmsf rmsf_ev.xvg -first 1 -last 1
如此,我们就得到了轨迹沿主成分1方向的两个极值投影,也即一个包含两帧结构的蛋白质坐标文件(ev.pdb),以及主成分值随原子号(残基号)变化的rmsf_ev.xvg数据文件。
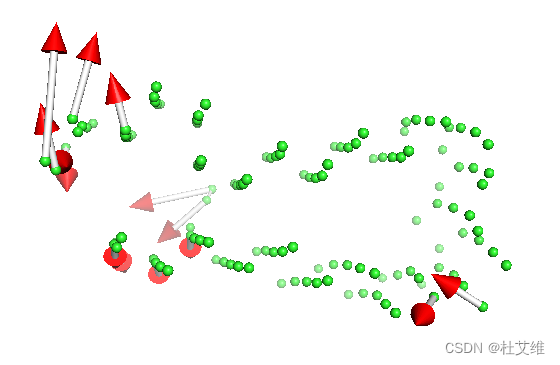
之后再把pdb文件加载到pymol中,用modevectors插件就可以观察到结构变化了:
modevectors ev11, ev11, 1, 2, factor=1, headrgb=(1,0,0),
tailrgb=(1,0,0), cutoff=0.5, outname=ev1
4. 其它DCCM绘制工具
除了著名的R语言包Bio3D,还有Python的一些库可以得到DCCM,如MD-TASK和Correlationplus。
MD-TASK
MD-TASK(https://md-task.readthedocs.io/en/latest/home.html)见刊于2017年,包含一系列python脚本,可以用于进行:
- Residue Interaction Network (RIN) analysis
- Perturbation Response Scanning (PRS)
- Dynamic Cross-Correlation
使用MD-TASK计算DCCM是简单的,下载github上的仓库,安装所需的依赖MDTraj和R语言的igraph等就行。执行如下命令计算DCCM:
python calc_correlation.py --prefix mdtask_res --trajectory md.xtc --topology md.pdb
需要给脚本提供一个轨迹文件和一个拓扑文件(可以用pdb文件),程序默认选择alpha-C进行DCCM的计算。
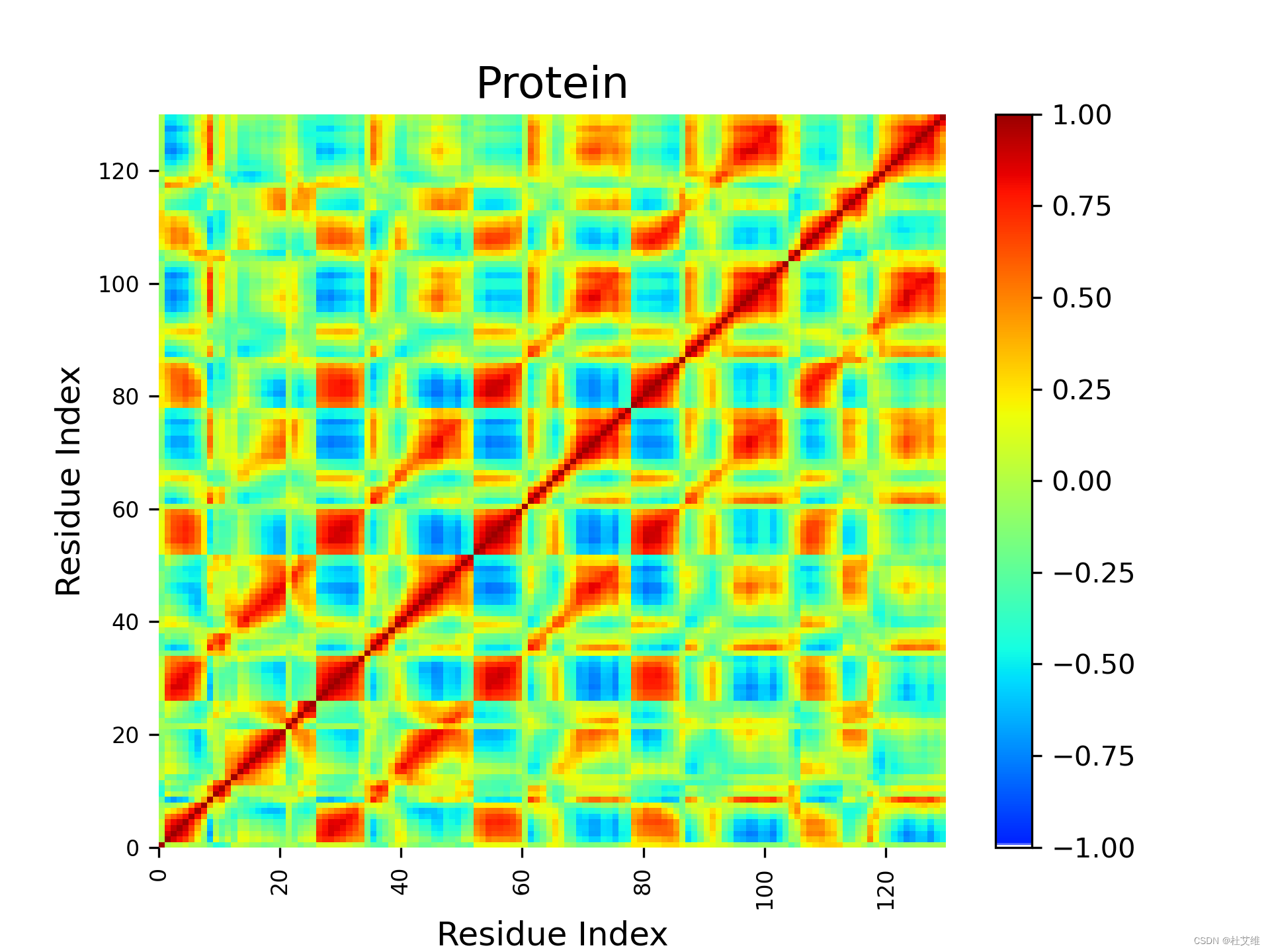
我用一个130个原子8002帧的体系进行了测试,需要14分钟才能计算完成,稍微有点儿慢。
程序会输出DCCM每一个点的数据到txt文件( N ∗ N N*N N∗N),也会直接生成一张DCCM图,如下:
Correlationplus
Correlationplus(https://github.com/tekpinar/correlationplus)见刊于2021年,也是一系列python脚本的集合,可以用于多种分析:
- normalized dynamical cross-correlations with Gaussian network model
- normalized dynamical cross-correlations with Anisotropic network model
- normalized dynamical cross-correlations from a molecular dynamics trajectory
- normalized linear mutual informations with Anisotropic network model
- normalized linear mutual informations from a molecular dynamics trajectory
- normalized Pearson cross-correlations of backbone omega dihedral angles from a molecular dynamics trajectory
- normalized Pearson cross-correlations of backbone phi dihedral angles from a molecular dynamics trajectory
- normalized Pearson cross-correlations of backbone psi dihedral angles from a molecular dynamics trajectory
Correlationplus被按功能分成多个模块,包括calculate、visualize、analyze等,每个模块又可用于多种分析和计算。下面简单阐述利用correlationplus计算DCCM的过程。
Calculate模块
执行命令如下:
correlationplus calculate -p md.pdb -f md.xtc -o corr_res.dat
得到的corr_res.dat就是最后的DCCM了( N ∗ N N*N N∗N)。 correlationplus的计算非常快,同样的体系,几秒钟。
Visualize模块
执行命令:
correlationplus visualize -i corr_res.dat -p md.pdb
执行结束之后会得到五个文件:
- 动态互相关矩阵图
- 互相关系数与残基距离的散点图
- 互相关系数与残基距离散点的数据文件
- 用于显示残基互相关的pymol脚本
- 用于显示残基互相关的vmd脚本(tcl)
先看动态互相关矩阵图:
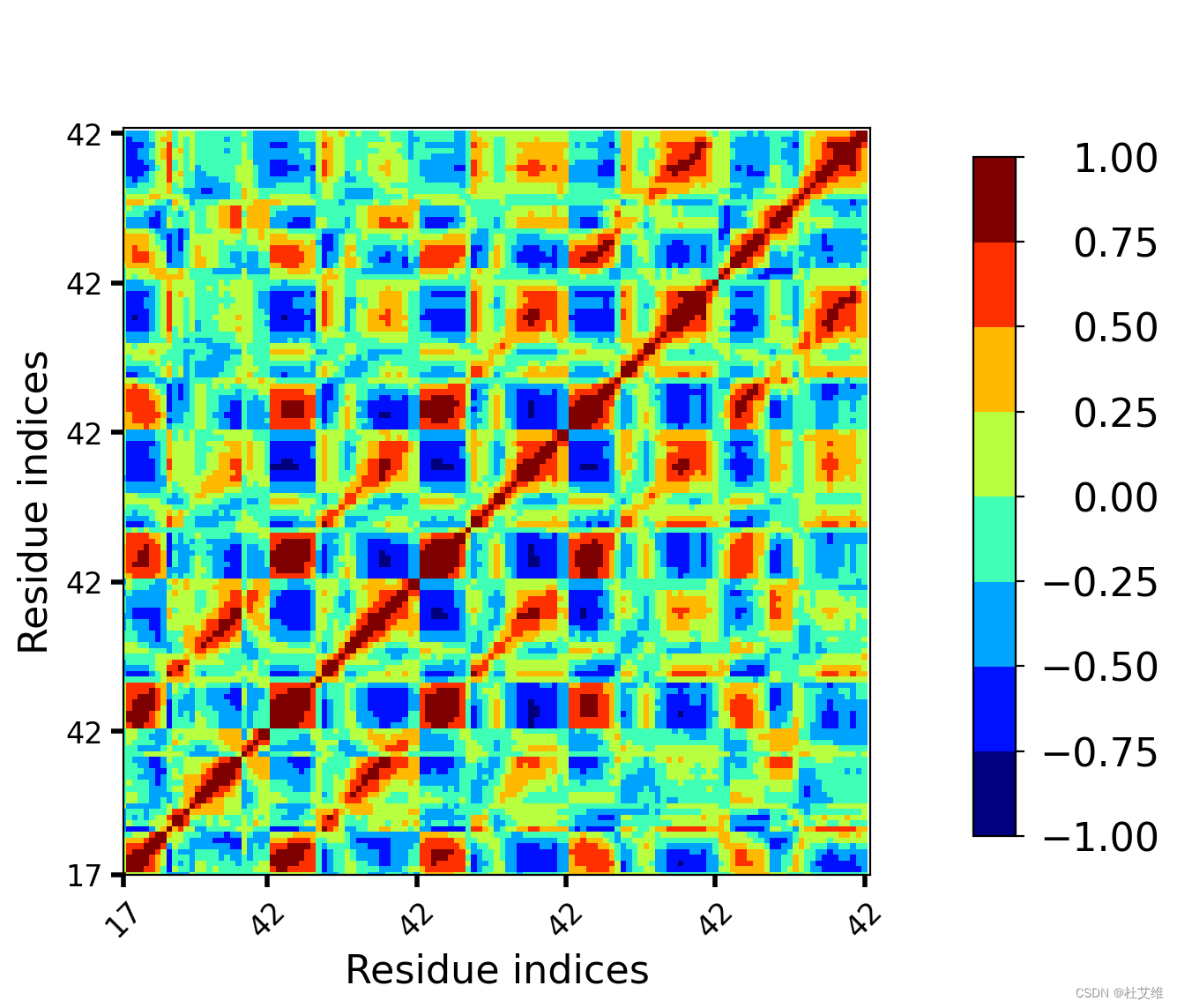
(我的蛋白质是个五聚体,Correlationplus没有对残基序号进行重编,因而如图。)
再看互相关指数与残基距离的散点图:
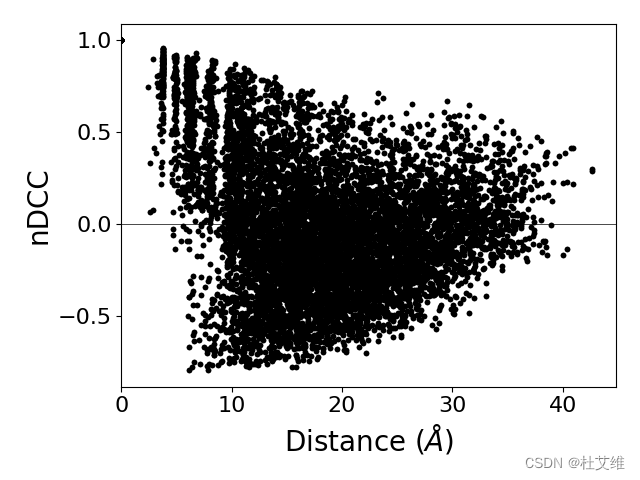
距离越远,相关性越小。
最后展示下通过pymol脚本显示出来的残基互相关结构图:
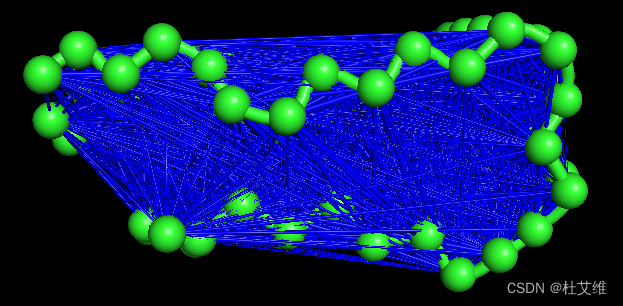
我这里绘制了所有的互相关指数,因而难看,可以通过-v和-x参数指定要绘制的互相关指数的范围,如绘制0.8到0.9的互相关指数:
correlationplus visualize -i corr_res.dat -p md.pdb -v 0.8 -x 0.9
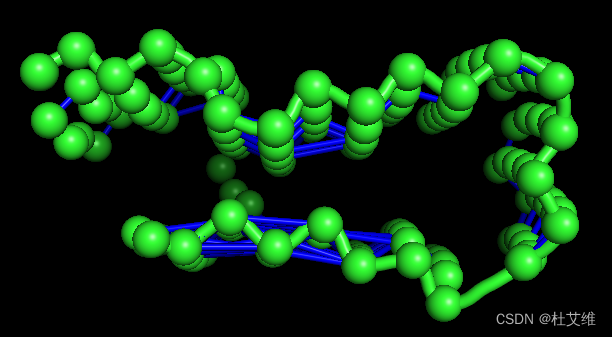
如此蓝线连接的就是互相关指数在0.8到0.9的残基了。
有一个值得小改进的地方,要是互相关的连接线能够按照互相关系数进行颜色的变化就更好了,目前线的粗细是和互相关系数关联的,但是辨识度确实不高。有空去PR哈哈哈。
diffMap命令
这个命令虽然官方文档没有着重讲,但也非常实用。我们经常需要比较不同情况下的DCCM图,比如说添加了配体的蛋白质DCCM图和无配体的蛋白质DCCM图。通过diffMap命令可以直接将两个互相关矩阵的数值对应相减,得到的就是DCCM的差异图。
比如说用结合了配体的蛋白质DCCM减去对照:
correlationplus diffMap -i pro_with_lig.dat -j control.dat -p md.pdb
得到图片:
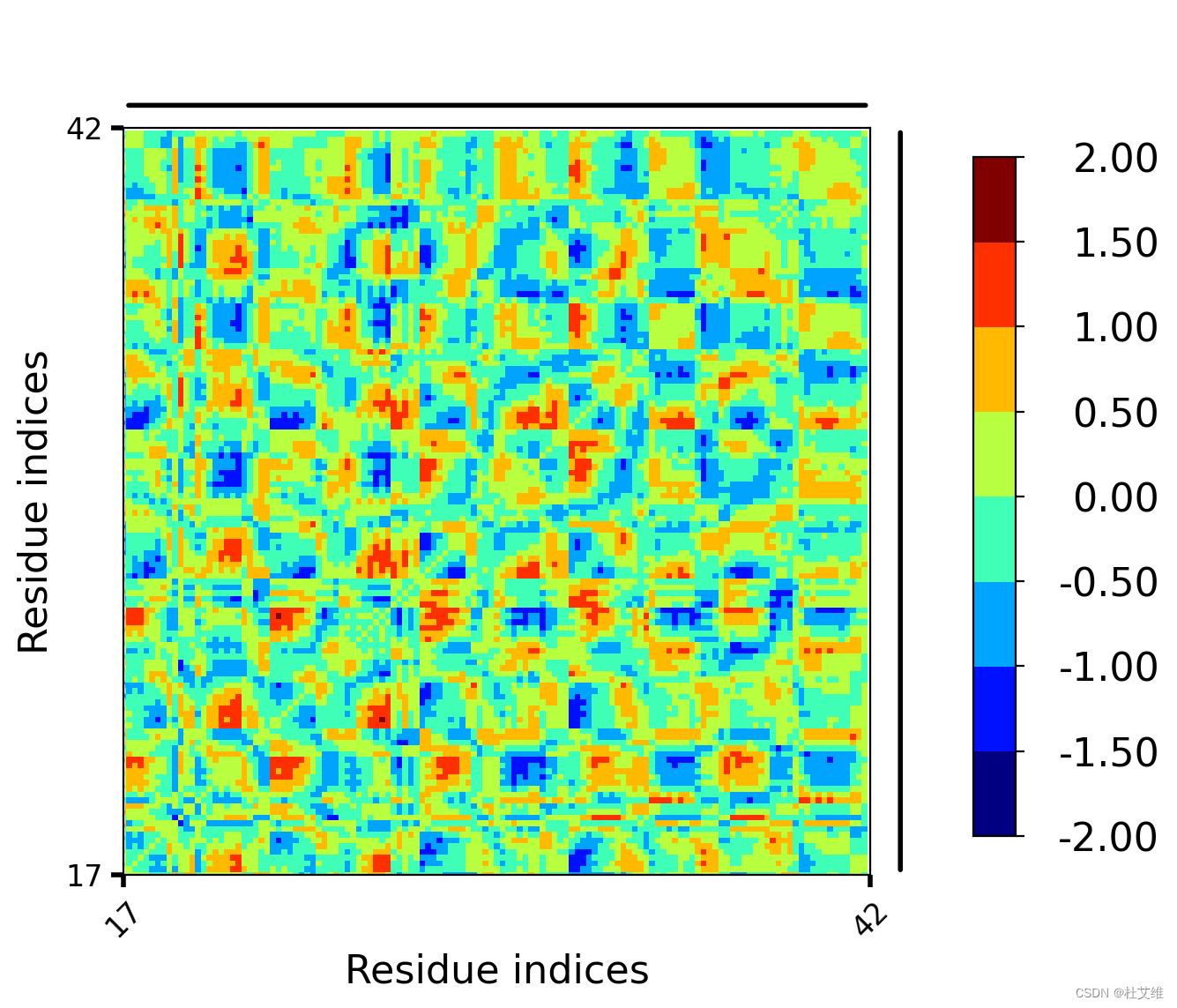
图上红色就表示:相较于对照组,带有配体的蛋白质的这部分残基间的相互作用更加正相关了;蓝色则表示相互作用更加负相关了。因而,配体对蛋白质的残基互相关的影响一目了然就能看到。
Analyze模块
analyze命令可以利用前面得到的数据进行中心性分析(centrality analysis),可以用于确定活性位点、结合位点、关键突变位点或者异构残基等。The script can compute degree, closeness, betweenness, current flow closeness, current flow betweenness, eigenvector centralities and major communities. 具体内容就留待大家自己探索啦~
5. 代码复现
理解了数学过程,就可以尝试自己写代码复现DCCM的计算和绘制。正好天大顾涛大佬也在尝试复现Bio3D的DCCM绘制,就曾一同交流讨论。感谢顾涛大佬在交流过程中分享的宝贵建议和经验哈哈哈哈哈,对代码感兴趣的可以移步大佬的repo:https://github.com/Zuttergutao/GMXAnalysis/blob/main/DCCM.py
点云fit
在计算协方差矩阵之前,很重要的一步就是对轨迹的每一帧结构进行对齐。不同的fit算法带来的结果是稍有差别的。
covar命令的最小二乘对齐:
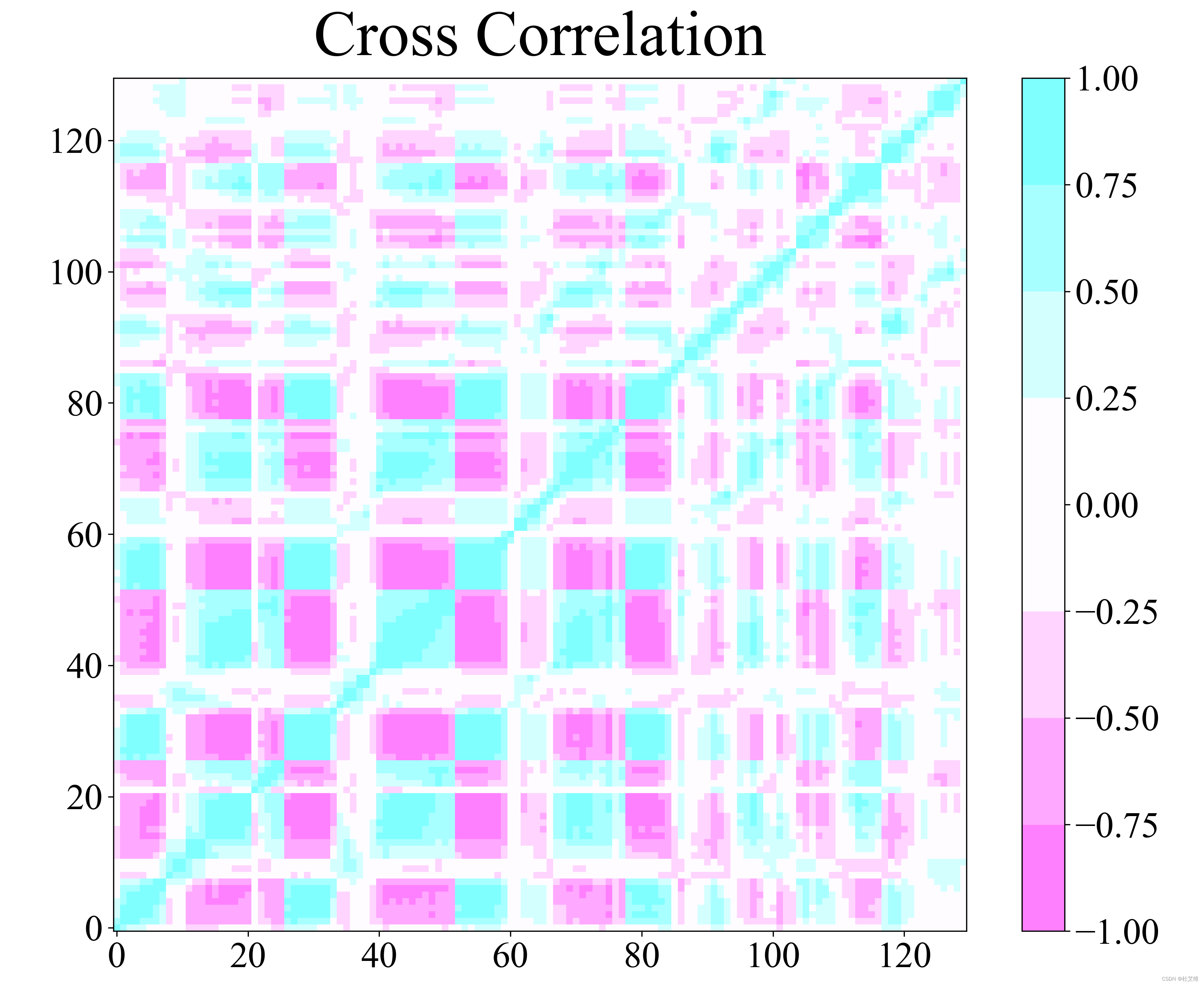
点云fit算法:
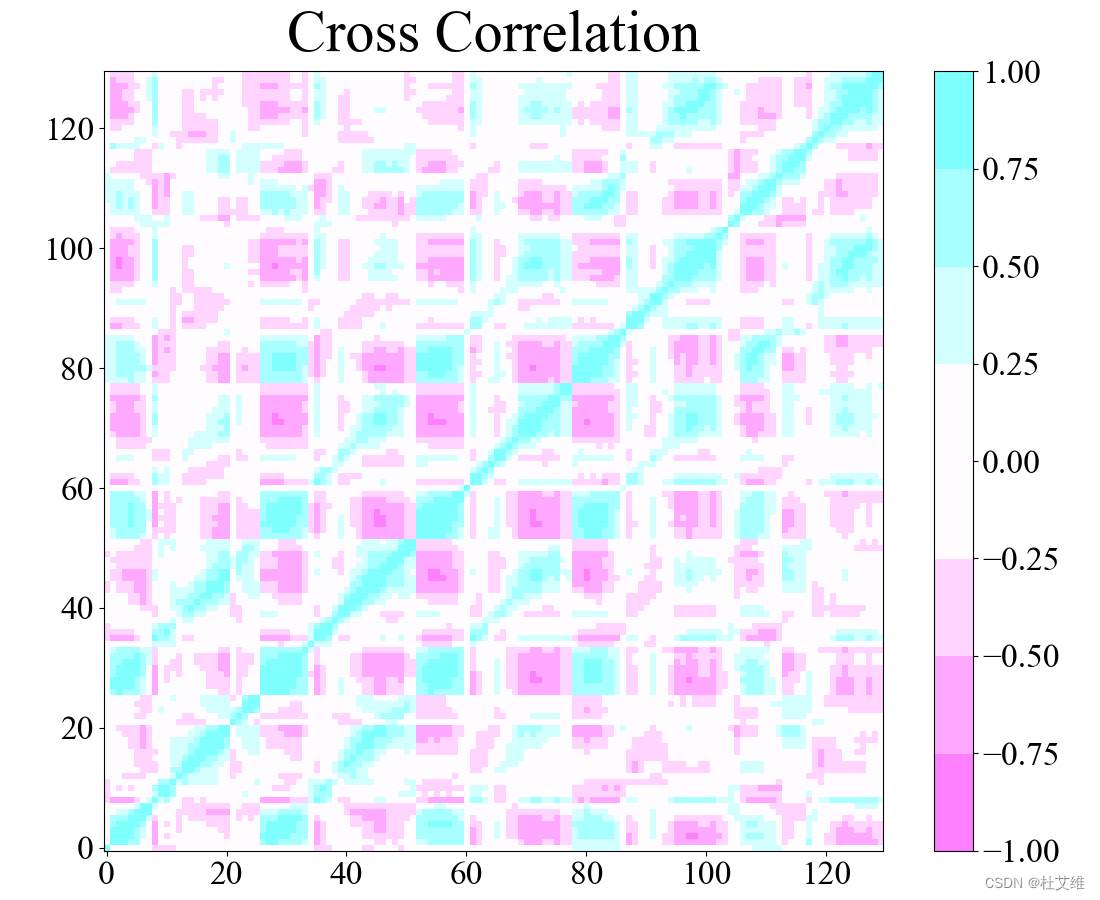
所以在计算不同体系的DCCM时,为了方便比较,最好使用同一种工具或者方法绘制DCCM,以避免不同fit算法带来的误差。
在复现过程中,我们使用了kabsch算法,可以参考https://zhuanlan.zhihu.com/p/535105203。
算法细节
总的计算流程是简单的。首先从轨迹文件中读取原子坐标,可以使用MDTraj或者libxtc等工具;然后使用对齐算法将每一帧的结构对齐到参考结构;之后计算所有帧的原子的平均位置,基于平均位置算出每一帧结构中每一个原子的位置偏移量;利用位置偏移量的两两点积算出每一帧的协方差矩阵,然后对时间求平均就得到最终的协方差矩阵了;最后对协方差矩阵的元素求相关性系数,就可以得到最终的动态互相关矩阵。
绘图细节
如果是使用matplotlib绘图的话,很多函数都可以做,比如说imshow()、matshow()、pcolormesh()、contourf()、contour()等,稍微调一下就会挺不错的。
要实现类似Bio3D的DCCM图的效果的话,可以尝试如下的绘图代码:
lis = ["#80FFFF","#A8FFFF","#D4FFFF","#FFFCFF", "#FFFCFF","#FFD4FF", "#FFA8FF", "#FF80FF"]
lis.reverse()
cm = matplotlib.colors.ListedColormap(lis)
im = plt.contourf(data, cmap=cm)
plt.contour(data, linewidths=0.2, levels=[-1.00, -0.75, -0.50, -0.25, 0.25, 0.50, 0.75, 1.00])
cb = plt.colorbar(im)
plt.xlabel("Residue No.")
plt.ylabel("Residue No.")
plt.title("Residue Cross Correlation")
plt.show()
contourf()形成等高线填充色块,然后用contour绘制一层等高线,效果如下:
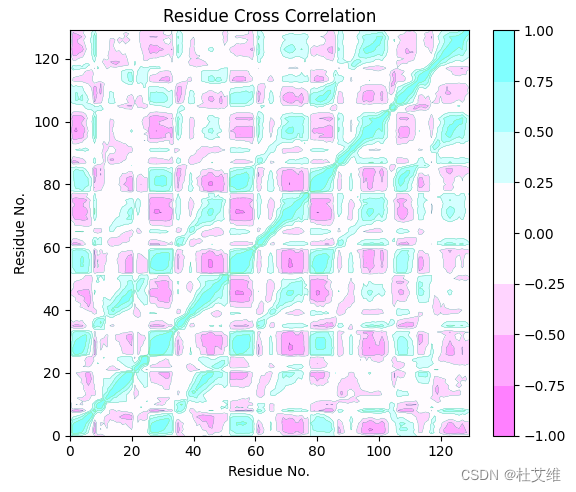
和通过Bio3D绘制得到的图基本上一致:
Others
诸位中秋节快乐!科研顺利!

















 4321
4321

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









