







1. 检查结构文件
有些结构文件存在少几个氢原子或者侧链的情况,所以先用spdbv软件打开结构文件,该软件可自动补加缺失的分子,用这个软件打开结构文件,再另存一下结构即可。
Spdbv软件是windows版本,可在网上直接搜索下载:http://www.genebee.msu.su/spdbv/text/getpc.htm
2. 准备参数文件
做动力学需要一些参数文件,具体文件中各项的内容还是看说明书吧,这个网址上有几个例子,这里拿第一个例子来做,该教程中提供了所有的参数文件http://www.mdtutorials.com/gmx/lysozyme/index.html
3. 在ubuntu中创建新的文件夹
为了整洁一些,做某个分子的动力学就建立一个文件夹,把参数文件和结构文件放里面,在里面做分子动力学,这样产生的所有文件就都在一起了,以免与其他文件混在一起,创建新文件夹和window系统一样,鼠标右键创建新文件夹即可。
【下面的步骤建议结合中英文教程一起看:
“https://jerkwin.github.io/9999/10/31/GROMACS中文教程/#1-水中的溶菌酶gmx-50“
“http://www.mdtutorials.com/gmx/lysozyme/01_pdb2gmx.html”
】
重要的地方是先检查pdb结构文件是否有问题,如是否缺失原子,这个结构是否是最佳结构等,如果不检查好,后续做完后才发现结构文件有问题就白忙乎了……这方面我吃过很多亏。
4. 生成拓扑文件(假设这里要做lysozyme.pdb的分子动力学,gromacs 5.0版本以上的软件都要在命令前加gmx,5.0以下的版本不加gmx,以下命令行均用红色字体表示)
gmx pdb2gmx -ignh -f lysozyme.pdb -o lysozyme_processed.gro-water spce
(-f是打开,-o为生成,这里需要选择一个力场,力场的选择可参照做此类分子的文献一般都选什么力场做,此处选择15)
5. 建立盒子
gmx editconf -f lysozyme_processed.gro -o lysozyme_newbox.gro -c -d 1.0 -bt cubic
6. 生成水盒子
gmx solvate -cp lysozyme_newbox.gro -cs spc216.gro -o lysozyme_solv.gro -p topol.top
7. 添加离子(该命令自动检测中和该结构所需要的电荷数,并自动添加na或者cl来中和)
gmx genion –s ions.tpr –o lysozyme_solv_ions.gro –neutral –p topol.top –pname NA –nname CL
选择溶剂组13SOL
8. 能量最小化
gmx grompp -f minim.mdp -c lysozyme_solv_ions.gro -p topol.top -o em.tpr
运行能量最小化
gmx mdrun -v -deffnm em
检测能量最小化情况
gmx energy -f em.edr -o potential.xvg
用grace软件打开potential.xvg文件查看能量是否平衡
xgrace potential.xvg
9. 能量最优化后首先保持蛋白质不动,对蛋白质周围水环境进行动力学模拟,该过程称为位置限制性分子动力学,对溶剂分子进行平衡计算,可以使溶剂分子填补空间:
gmx grompp -f nvt.mdp -c em.gro -p topol.top -o nvt.tpr
运行:
gmx mdrun -deffnm nvt
检测温度是否平衡
gmx energy -f nvt.edr -o temperature.xvg
10. 平衡压力
gmx grompp -f npt.mdp -c nvt.gro -t nvt.cpt -p topol.top -o npt.tpr
运行:
gmx mdrun -deffnm npt
检测压力和密度情况
gmx energy -f npt.edr -o pressure.xvg
gmx energy -f npt.edr -o density.xvg
11. 分子模拟
gmx grompp -f md.mdp -c npt.gro -t npt.cpt -p topol.top -o md_0_1.tpr
运行:
gmx mdrun -deffnm md_0_1
如果大家已做完前面的步骤,得到了模拟后的文件,就可以继续对结果进行分析,采取什么方法分析取决于你想解决什么样的问题,是比较野生型与突变体结构的差别还是分析底物与受体之间的作用力,这里列举几个我常用的方法,希望对大家能有帮助。
gmx trjconv -s md_0_1.tpr -f md_0_1.xtc -o md_0_1_noPBC.xtc -pbc mol-ur compact
选择0:system用于输出,基于这个“修正”后的轨迹进行分析。
1. 生成某时间段的平均结构(用-b, -e限制时间,如-b 1000 -e 2000指:从第1000ps开始到2000ps)
gmxcovar -f md_0_1.xtc -s md_0_1.tpr –b 1000 -e 2000 -av traj_avg.pdb
2. 统计某时间段的氢键数目(用-b, -e限制时间)
gmxhbond -s md_0_1.tpr -f md_0_1_noPBC.xtc -b 25 -e 40 -num hnum.xvg -tu ns
3. 盐桥(用-b, -e限制时间)
gmxsaltbr -s md_0_1.tpr -f md_0_1_noPBC.xtc -b 39900 -e 40000 -t 10000
4. 将模拟后的.gro文件转化为.pdb文件
gmxeditconf -f md_0_1.gro -o md_0_1.pdb
5. 两个结构叠加
gmx confrms-f1 model1.pdb -f2 model2.pdb -o fit.pdb
我一般是用pymol软件简直对两个结构叠加。
6. do_dssp使用
安装:
a.在其官网上下载安装包http://swift.cmbi.ru.nl/gv/dssp/ 下载dssp-2.0.4-linux-and64
b. 将安装包移动到目录/usr/local/bin下面
sudocp dssp-2.0.4-linux-and64 /usr/local/bin
c. 进入/usr/local/bin
cd/usr/local/bin
d. 文件重命名 mv dssp-2.0.4-linux-and64 dssp
c. 修改权限 chmod a+x /usr/local/bin/dssp
e. 在工作目录下面输入命令 export DSSP=’usr/local/bin’
运行:
gmxdo_dssp -s md_0_1.tpr -f md_0_1_noPBC.xtc -tu ns
gmxxpm2ps -f ss.xpm -bx 0.1 -by 4 -o ss.eps
(注:可以修改-bx -by后面的参数改横纵坐标的长度,ss.eps文件可用ps打开,另存为普通照片格式的图片)

7. 计算残基之间的最小距离
(1) 先用make_ndx命令生成index文件:
gmxmake_ndx -f md_0_1.tpr -o index.ndx
(2) 这里选择某蛋白第11个氨基酸的H原子和36位氨基酸的O的距离:
选择原子:r 11 & a H 回车 r36 & a O 回车 q(退出)
(3) 用mindist计算最小距离:
gmxmindist -f md_0_1_noPBC.xtc -s md_0_1.tpr -n index.ndx -od dist.xvgr
选择刚刚选择的两个原子所在的组,如两者分别在第18和19组的话进行如下操作:
18回车19回车 按ctrl-D退出
(4) 作图
生成的dist.xvgr文件可用excel作图,横坐标是时间,纵坐标是距离
8. 详细的作用力分析
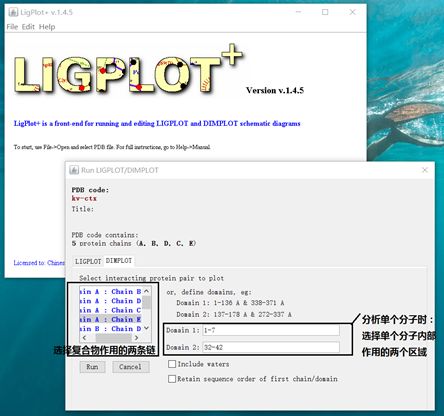
可用LigPlot+软件分析复合物之间或者单个分子内部的作用力
LigPlot+软件官网:http://www.ebi.ac.uk/thornton-srv/software/LigPlus/
下载的安装包直接放到c盘解压即可,这里需要电脑安装java,如果没有的话软件打不开。
使用:打开Ligplot软件,如果是复合物,需要限定作用的侧链,如果是蛋白内氨基酸之间作用力分析,需要写作用的两个domain的氨基酸序号,然后点击run即可。
最后,有分子动力学模拟相关需求欢迎通过微信公众号联系我们。
微信公众号:320科技工作室。
同时,欢迎大家关注微信公众号“Becky的生活and科研”。
















 671
671











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











