







参考:
【1】http://www.bioengx.com/bio3d%e7%9a%84%e5%ae%89%e8%a3%85%e4%b8%8e%e5%ba%94%e7%94%a8%e5%ae%9e%e4%be%8b%ef%bc%88dccm%ef%bc%89/
【2】http://thegrantlab.org/bio3d_v2/user-guide
【3】http://thegrantlab.org/bio3d_v2/tutorials/protein-structure-networks
1.Bio3D的介绍
Bio3D 是一款依托于 R 语言的工具包,包含用于分析生物分子结构、序列和轨迹数据的实用程序。功能包括读取和写入生物分子结构、序列和动态轨迹数据的能力,执行原子选择、重新定向、叠加、刚性核识别、聚类、距离矩阵分析、守恒分析、正常模式分析和主成分分析。
Bio3D应用实例
以Bio3D计算DCCM作为应用实例。
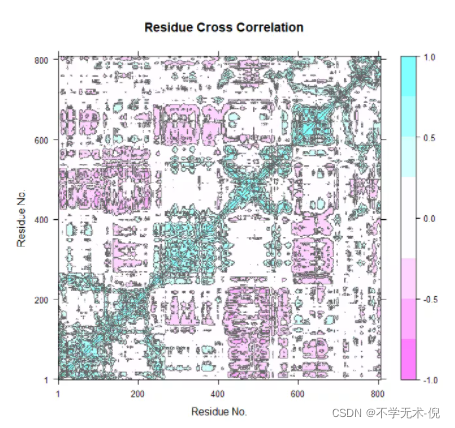
动态相关性矩阵(Dynamic cross-correlation matrices, DCCM),表示蛋白质中每个氨基酸的特定原子,比如Cα原子和其它氨基酸的Cα原子之间的相关性,提供蛋白质在大尺度范围内相关运动的一些信息。DCCM计算数值的取值范围从完全负相关的-1.0到完全正相关的+1.0。越接近数值1表示相关性越强,正负表示两个原子运动方向相同(反)。
首先需要准备两个文件.dcd和.pdb(以taq DNA聚合酶为例)

library(bio3d)
require(bio3d); require(graphics);
pause <- function()
cat(“Press ENTER/RETURN/NEWLINE to continue.”)
pause()
trtfile <- system.file
(“D:/Program Files/R/R-4.1.0/bin/x64/start.dcd”,package=”bio3d”)
dcd <- read.dcd(“D:/Program Files/R/R-4.1.0/bin/x64/start.dcd”) (#定位到.dcd和.pdb所在的文件夹)
pdbfile <- system.file
(“D:/ProgramFiles/R/R-4.1.0/bin/x64/start.pdb”,package=”bio3d”)
pdb <- read.pdb(“D:/ProgramFiles/R/R-4.1.0/bin/x64/start.pdb “)
print(pdb)
pause()
dim(dcd)
ncol(dcd) == length(pdb$xyz)
pause ()
ca.inds <- atom.select(pdb, elety = “CA”)
xyz <- fit.xyz(fixed = pdb$xyz, mobile = dcd,
fixed.inds = ca.inds$xyz,
mobile.inds = ca.inds$xyz)
DCCM的计算:
cij <- dccm(xyz[,ca.inds$xyz])
plot(cij)
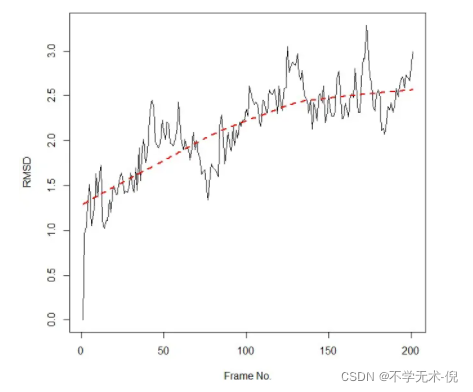
RMSD的计算(表示整个结构随时间的位置差异):
rd <- rmsd(xyz[1,ca.inds$xyz], xyz[, ca.inds$xyz])
plot(rd, typ= “l”, ylab = “RMSD”, xlab = “Frame No.”)
points(lowess(rd), typ =”l”, col = “red”, lty = 2, lwd = 2)
summary(rd)
pause()
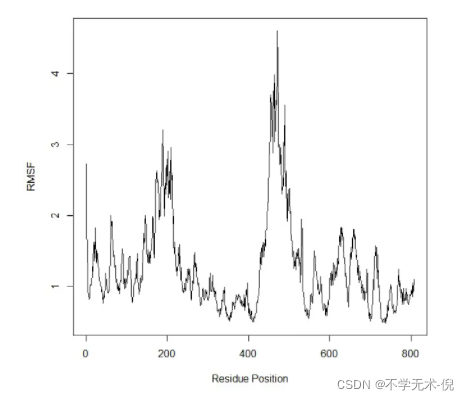
RMSF的计算 (表示单个氨基酸残基的柔度,或计算模拟过程中特定残基移动的程度):
rf <-rmsf(xyz[, ca.inds$xyz])
plot(rf, ylab =”RMSF”, xlab = “Residue Position”, typ=”l”)
pause()
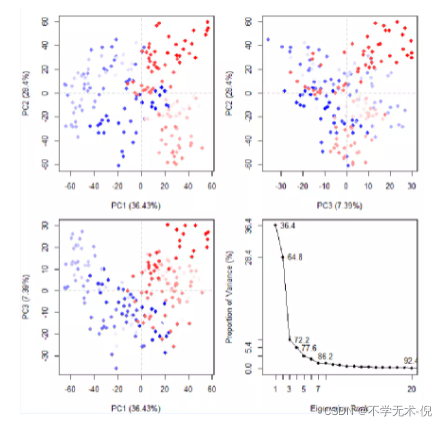
PCA的计算(Principal ComponentAnalysis,主成分分析)
pc <- pca.xyz(xyz[,ca.inds$xyz])
plot(pc, col= bwr.colors(nrow(xyz)))
备注:.dcd文件可以通过VMD来获得。即
先将GROMACS得到的last10ns.xtc转为start.pdb
gmxtrjconv -s md.tpr -f last10ns.xtc -o start.pdb -dump 0 (选择1, protein)
打开VMD,将start.pdb转存为start.dcd














 2177
2177











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









