









参考https://github.com/MaybeBio/Chemoinformatics/tree/main/pymol/CTCF_ZF1-7
脚本是:
主要教程参考:
http://pymol.chenzhaoqiang.com/intro/advanceManual.html
仅剩这2部分还未自学
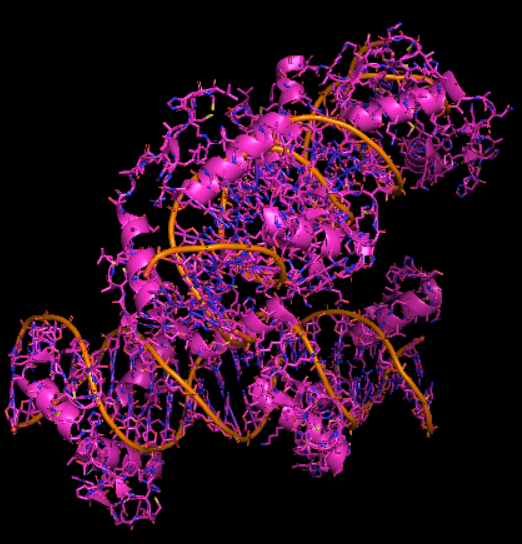
Q:自选一个pdb,使用pymol做图说明该生物大分子(蛋白)的功能、特征或催化机制等。
用图注详细解释图片的内容。需要包括图和图注,如下图:


pymol下载linux版,使用conda下载:

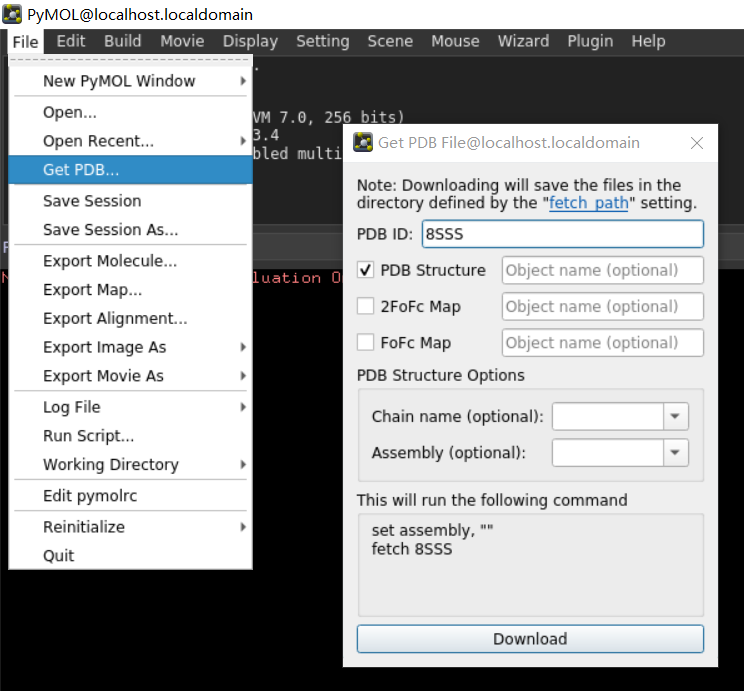
1,获取PDB,可以自己在PDB数据库上下载然后导入pymol,或者是直接在pymol软件上下载对应数据:
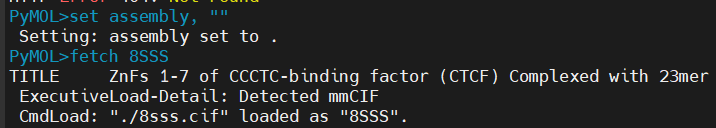
#1,打开日志,记录后续所有的操作指令:
#打开XXX日志,也就是xxx.pml
log_open CTCF_ZFmotif_1.pml
#注意在最后拍完照结束的时候
log_close
#2,调整工作目录:

#3,加载蛋白PDB文件:

实际上的命令行操作:
cd /mnt/sdb/zht
#加载PDB蛋白,以8SSS为例
set assembly,""
fetch 8sss
4,PDB中对对象(例如蛋白质、DNA的)简单操作:
(1)蛋白的展现形式:
每一个object的A/S/H/L/C操作

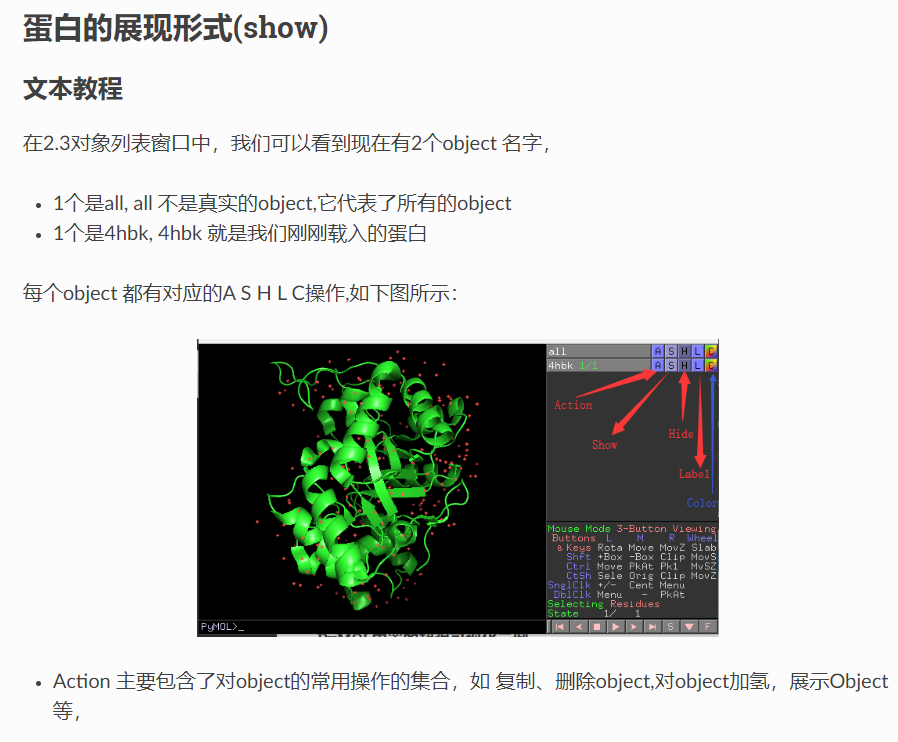

事实上我们知道:所谓的as只是蛋白质的展示方式
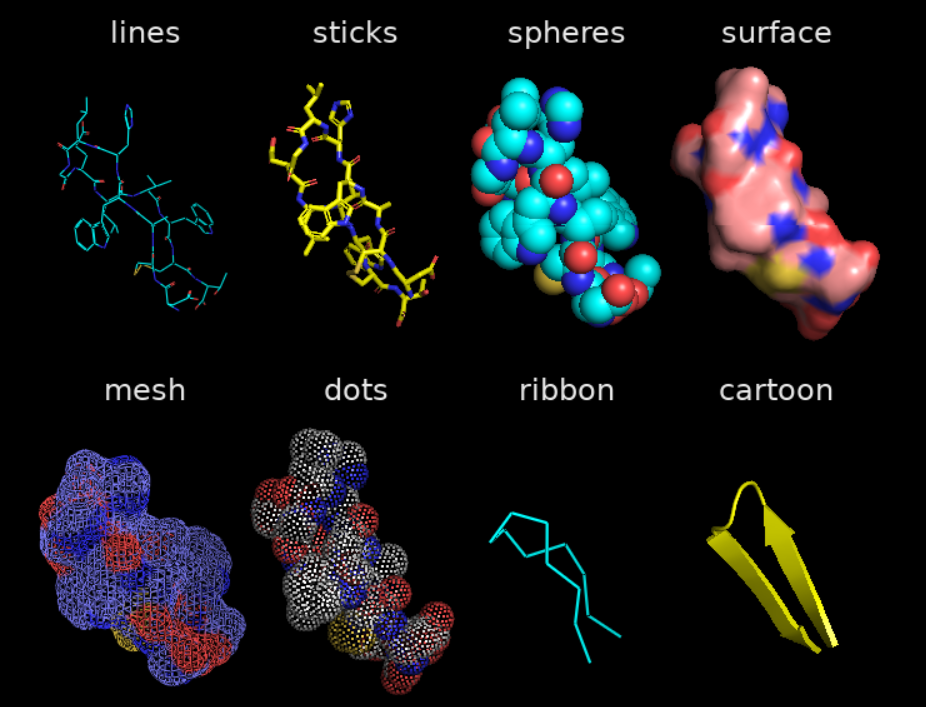

所以上述show——》as的展示差异只是渲染效果(渲染成生化中的什么化学结构式)有差异罢了,一个是两种渲染效果不兼容,只能保留最后最新一种,一种是能同时渲染多种效果
show是嵌套多种显示效果,as是只保留最后一种效果
**总之,一般情况下就用show展示效果,比如说要展示CTCF的ZF和DNA上的motif碱基之间的具体互作,以及测量距离,原本的对象是cartoon,此时为了美观要统一效果就as sticks(可以通过撤回取消);
之后测量完距离,要做真空静电力学的时候就使用show surface,然后做完了surface之后,要对下一个ZF做互作展示+测量distance了,就hide surface然后对原本的stick做dist。**
as可以通过撤回取消,或者是重新选择对象之后使用原本的效果as
show的效果除了撤回,还可以通过hide隐藏(比如说show surface,之后hide surface)
 撤回键
撤回键
(2)蛋白对象的平面操作

平移:ctrl+左键,或者是鼠标滚轮中键一直按
旋转:仅仅左键
缩放:仅仅右键,下拉放大,上移缩小
切割(剖面):仅仅鼠标中键
如果涉及到复杂的角度操作:
rock:绕着竖直轴旋转,也就是将正面以及反面转向我们


(3)蛋白质-DNA复合体的对象分离:select以及create
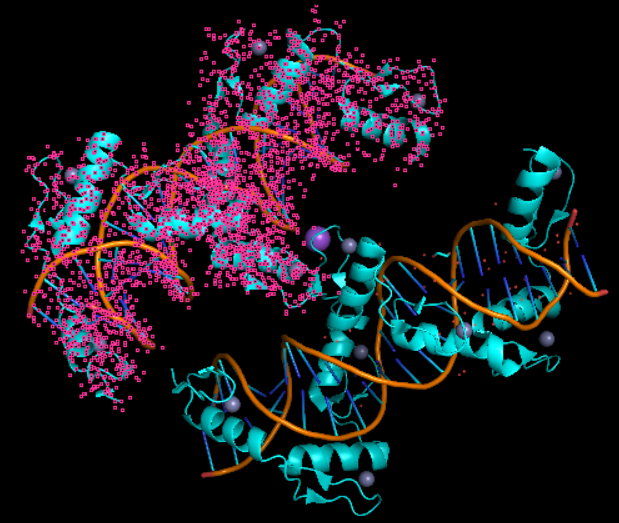
一般需要通过PDB网站查询该晶体结构的组成,
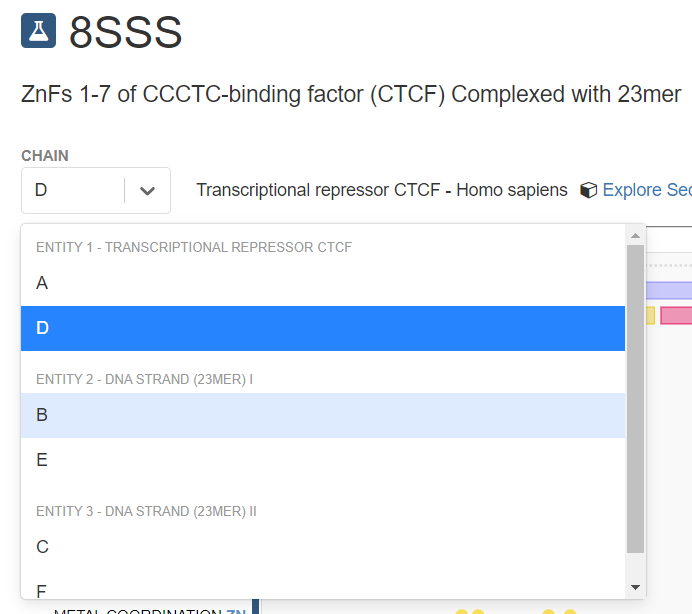
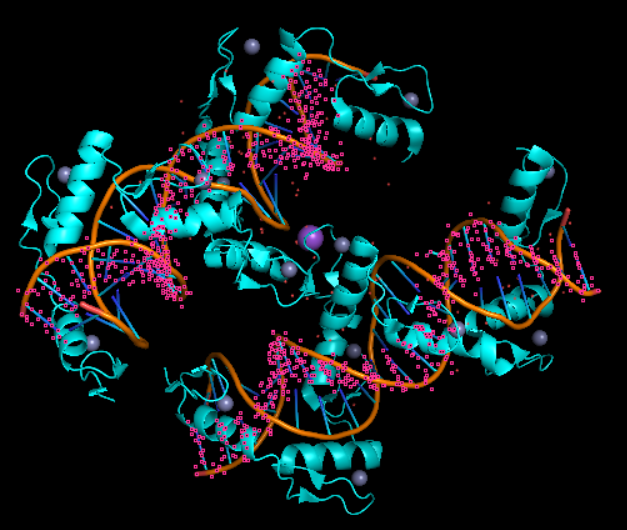
查看结构:发现是二聚体
只需选取其中蛋白质A,DNA B+E即可
首先试验一下:
A/D是蛋白质,即CTCF的ZF1-7

然后B/E是双链DNA,连接在链A上

也不对,应该是按照序号来的,所以应该是
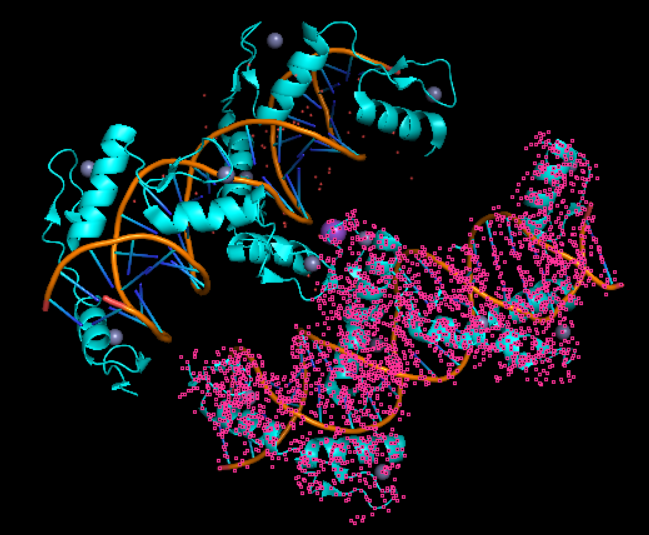
蛋白质A+双链DNA B+C
蛋白质D+双链DNA E+F
——》总结就是:
在普通操作上,用的最多的命令就是select、create,其余的基本都是移动角度的指令
select可以指定对象范围(在命令行中),也可以直接在pymol GUI中鼠标左键一个一个点击然后选择sele
注意各种生物大分子选取对象的命令:
虽然从逻辑上来看下面选择的操作指令不一定科学严谨,但是从效果上来说是合理的,
另外注意各种符合之间的等价,&与and,CA或者P等大小写不敏感;
另外就是各种and之间是缩小定义对象范围,而or是扩大范围;
另外就是要注意我么选取的对象是以什么单位为基础进行选择的:
是每一个原子(这样label的时候每一个原子都会有label ZFXX),还是以残基,还是以聚合物等等
实际上以什么最小单位选取对象也是会影响选取对象的准确性以及之后操作的严谨性的

(4)label操作:
基本上所有的指令都涉及到:指令+对象(范围)+参数等
label主要需要关注的是标注的内容的修改:也就是label内容的修改

主要是字体等、阴影等、轮廓等
5,选取对象+展示好效果+选对角度之后进行拍照:
(1)如何选取好角度:
对于蛋白质的平面操作见前所述4(2)蛋白对象的平面操作,主要就是平移+旋转+缩放等;
可以迁移到DNA中
(2)

(3)定好之后拍照:
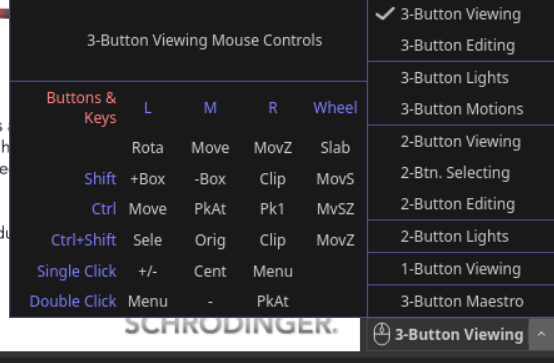
#基本上对象+角度定下来之后,拍照细节如下:
bg_color white
ray 1000,1000
#此时可以考虑将label的字体以及展示效果进行修改
set label_size xxx
#以及通过3-button editing拖动展示残基:ctrl+左键
#保存图片
png xxx.png
6,其他:
(1)脚本类型:
pse是对话,也就是保存当前任务中所有信息
pml是脚本,也就是记录终端中任何命令行+鼠标操作(相当于是log日志,我会在任务一开始时记录,也会将脚本记录在pml中)
py脚本也兼容,但是在语法上需要注意点:
http://pymol.chenzhaoqiang.com/intro/advanceManual.html
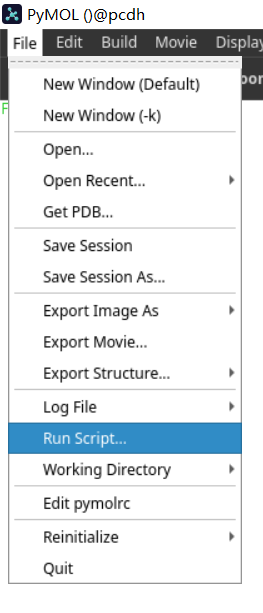
主要就是python脚本如何在命令语法上与pml脚本相互兼容:
pml脚本实际上就相当于是pymol的终端,
其实主要是
#在python脚本中一定要使用cmd:
from pymol import cmd
#也有些脚本会额外加载其他ku
from pymol import cmd, util
#然后python脚本中的一切命令需要使用cmd.xxxx
#当然在pml脚本中也可以直接使用cmd.XXXX(在pymol终端中不需要import库)
我一开始是使用pml脚本为主,但是后来发现在pml脚本中的指令本质上就是pymol终端中输入的指令,
所有一些python脚本的限制,在终端中被限制的在pml脚本中也会被限制:
例如:我在python脚本中会定义一些函数模块,使用def xxx(yyy):
我们知道函数模块会有缩进要求,但是在pymol终端中输入的时候无法识别会报错,那自然放在pml脚本中就也会报错,所以实际上如果想要python脚本的完全自动化,就最好不要在pml脚本中混杂python代码,
而是直接全部all in python脚本,使用cmd等进行编写——》建议所有pymol脚本直接写在py中



(2)如何撤销undo:
一般是展示效果和对象上:首先试试撤回
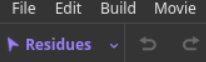
实际上在终端中ctrl+Z也可以达到撤回效果,ctrl+Y可以达到恢复效果
选错了某个对象,或者是创建失误:可以delete对象之后redo
展示效果上的失误:
如果是show,可以hide:show surface——》hide surface
如果是as,暂时只能撤回,或者重新选择对象之后as回来原本的展示效果(所以建议不要全局as,一切操作最好都是针对具体范围对象,这样as回来的时候也就可以针对性as,如果一开始是全局as,那纠正撤回的时候就不清楚范围了)
如果是真空静电力学的效果展示:
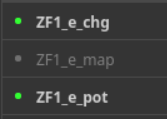
只要将这3个对象hide即可;
如果想撤回的操作范围过大无法判断:只能delete all,然后将原本log_open脚本中的命令再一条一条粘贴回来重新redo
(3)临时保存对象:
如果任务执行到一半,没有结束,需要下一次继续执行,就必须等保存当前所有的会话中的对象,
相当于R中的.rda (也称为.RData )文件,
主要是:
#保存当前会话,用于下一次继续
save xxx.pse #建议使用当前时间来记录会话,然后下次就是在file——》open
#保存对象,如果对会话中过程性对象不需要的话
save xx.pdb
save xx.pdb,yy对象
#前者是保存全部对象,后者是保存指定对象
(4)其他pymol相关的python库:
结合RDkit:
conda安装https://github.com/rdkit/rdkit
7,回归到本项目:
首先是数据上:
ZF1-ZF7+oligo 23 bp:8SSS
ZF3-ZF11+oligo 35-4:8SSQ
ZF3-ZF11+oligo 35-20:8SSR
ZF3-ZF10+oligo 19:8SSU
暂时只获取8SSS作为后续数据处理对象,


以下为絮絮叨叨执行草稿部分
(1)首先是打开log日志文件:
log_open 这个session名字.pml
(2)标注出不同的ZF序列
给一个蛋白质标注不同的part:
那首先要从中分离出来蛋白质和DNA部分:
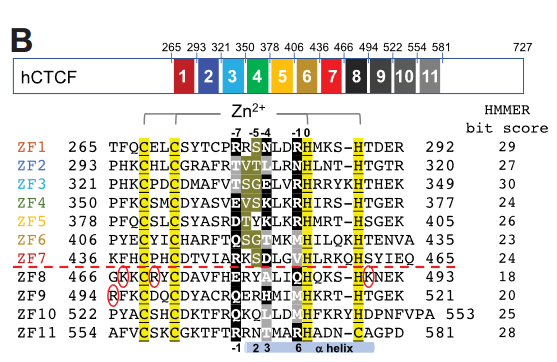
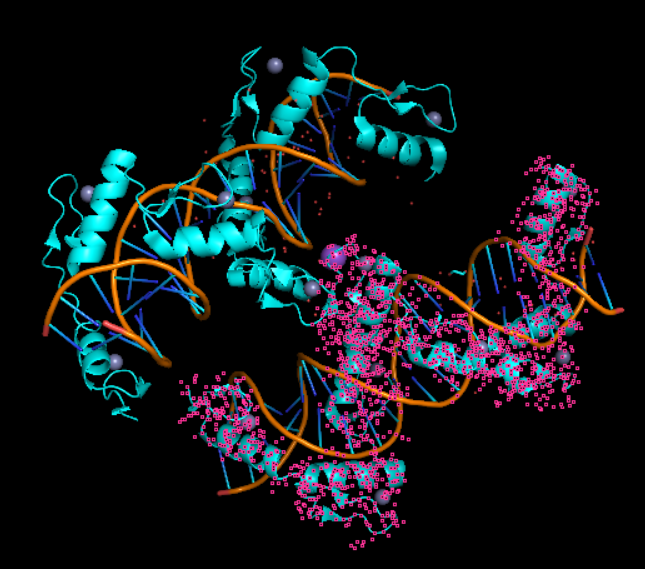
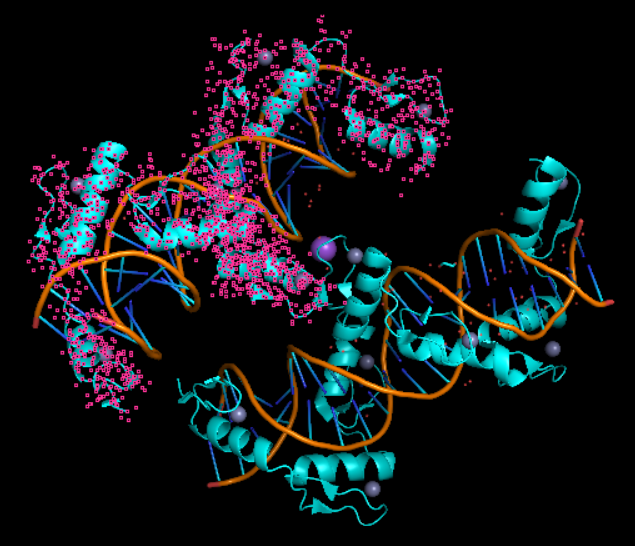
查看结构:发现是二聚体
只需选取其中蛋白质A,DNA B+E即可
首先试验一下:
A/D是蛋白质,即CTCF的ZF1-7
然后B/E是双链DNA,连接在链A上
也不对,应该是按照序号来的,所以应该是
蛋白质A+双链DNA B+C
蛋白质D+双链DNA E+F
接下来使用一个单体进行操作分析
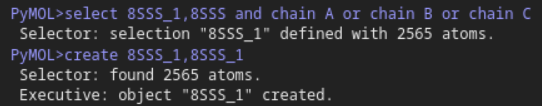
接下来的分析需求可以参照文献中的图:
按照序列来选择:
我的建议是先分离蛋白质和DNA,也就是ZF1_7与ZF1_7_motif
再然后对ZF1_7依据序列数据标明1-7
首先是蛋白质的分离:
接着是DNA:
然后再ZF1_7上面依据氨基酸残基序列标注不同的ZF片段
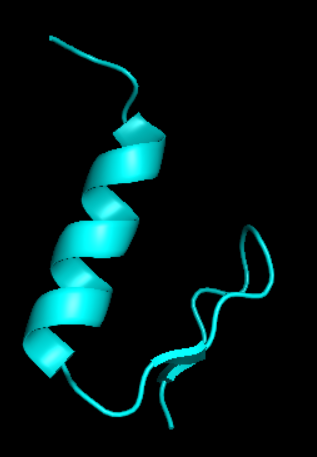
同理完成ZF1-7的标注
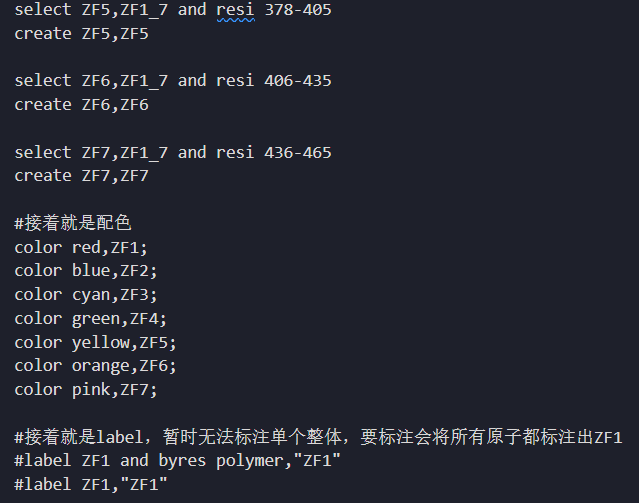
上面是分开做的事情,当然还有合起来的在整体上也用不同颜色label不同的ZF
首先是依据resi的数据在ZF1_7上分段color,再然后label
因为color以及label都是要对同一对象进行,所以统一选择select
但是label的时候应为一开始选取的是所有残基的集合,所以label的时候是集合中的所有原子都有ZF1 label,而不是只有一个label;
所以建议还是在select之后create,
也不行,建议不用在select之后再分离create对象
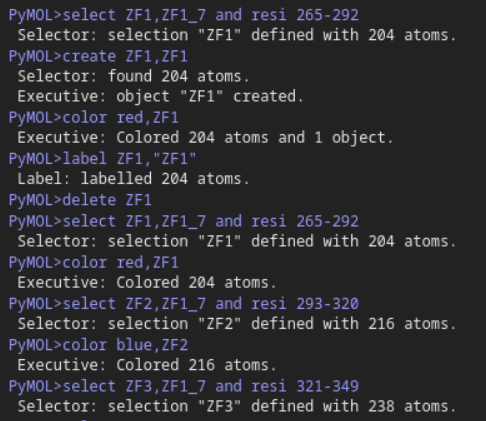
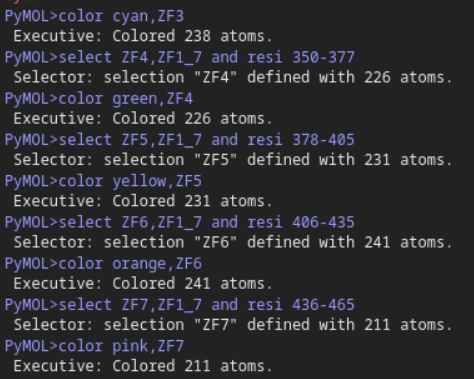
总之处理之后:
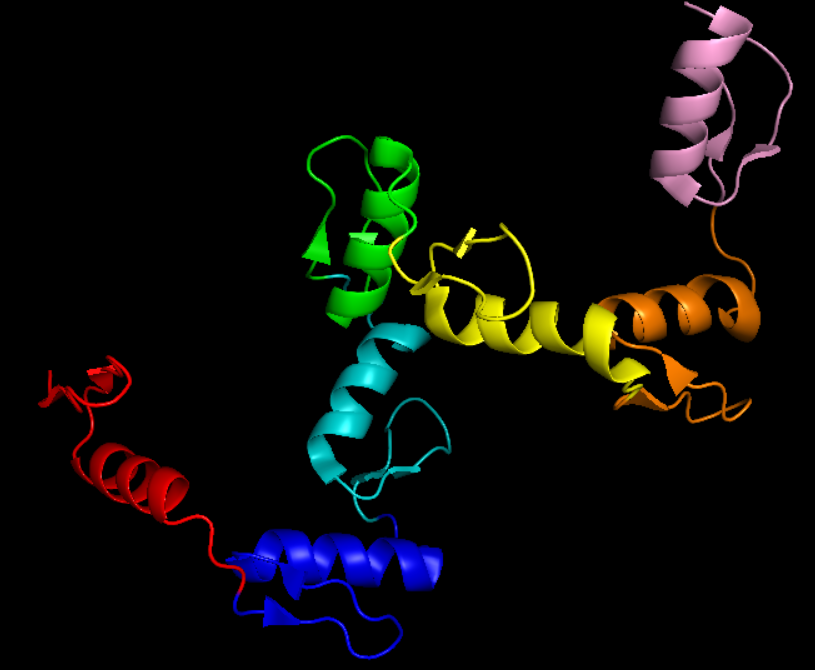

(3)其次是要在不同的ZF序列中找到对应的序列特异性位点,可以选出来,也可以直接在整条序列中label出来
如何识别两条互作序列中的互作位点
或者是位点已知:
选取其中的结晶链


具体自动化label的话,也是和开始一样,先选中氨基酸还是DNA,然后使用占位符
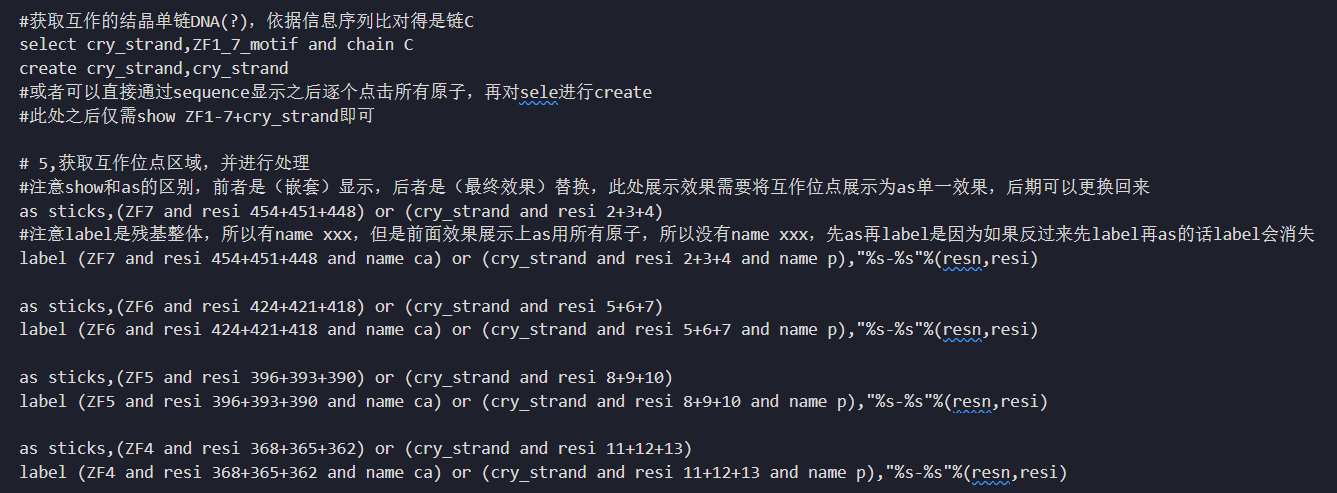
再create一个ZF7,也就是互作的对象要分离,然后互作的部分要select选中再as sticks
或者直接鼠标点击选中几个互作残基,然后sele上进行show,注意要as(如何只展示一种效果)
从文献中获取已知信息:
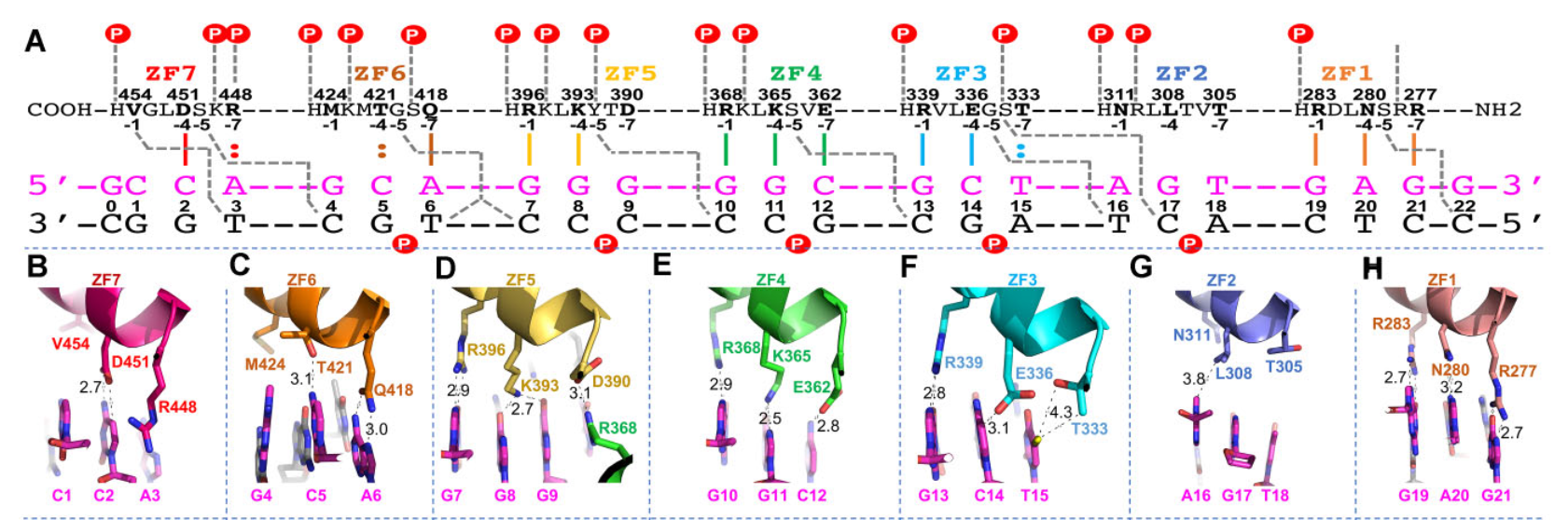
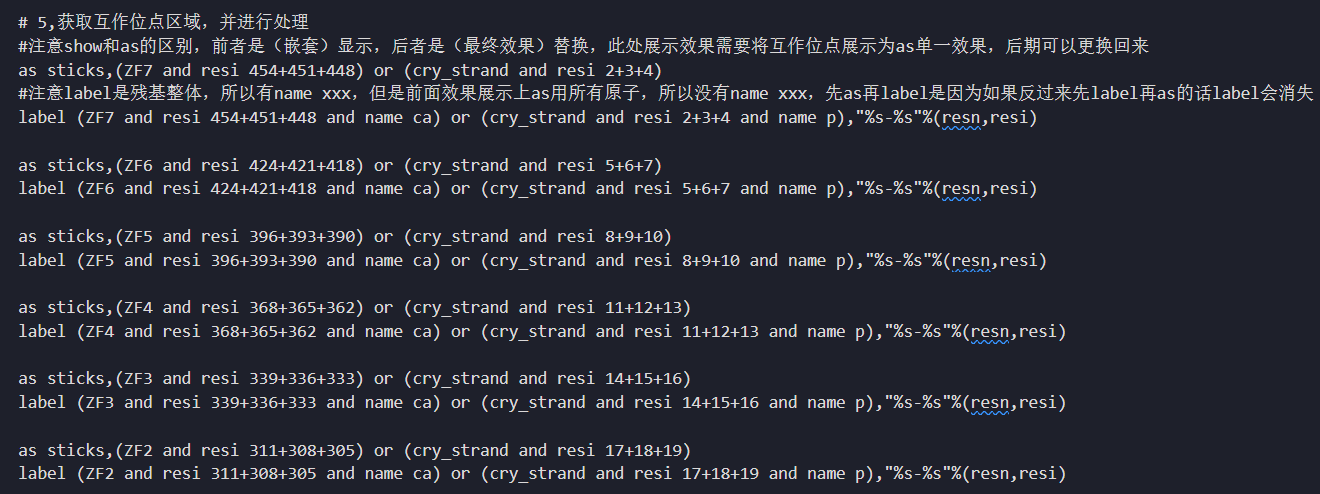
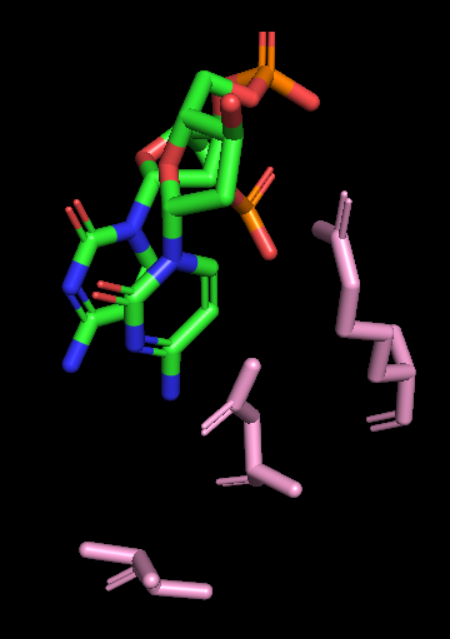
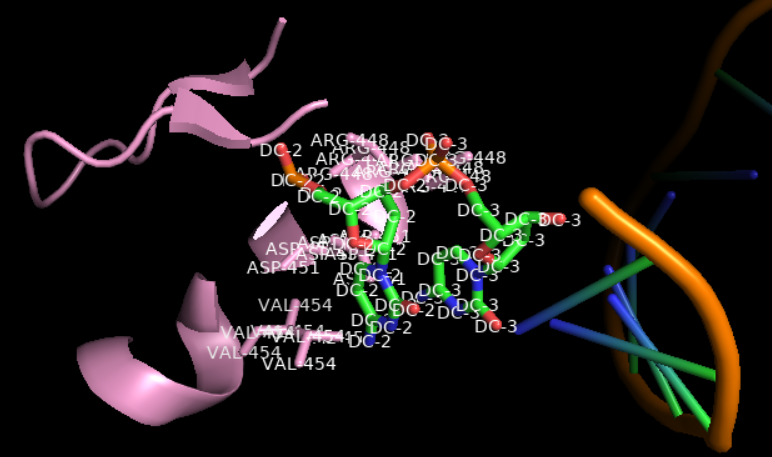
ZF1_7
ZF7:
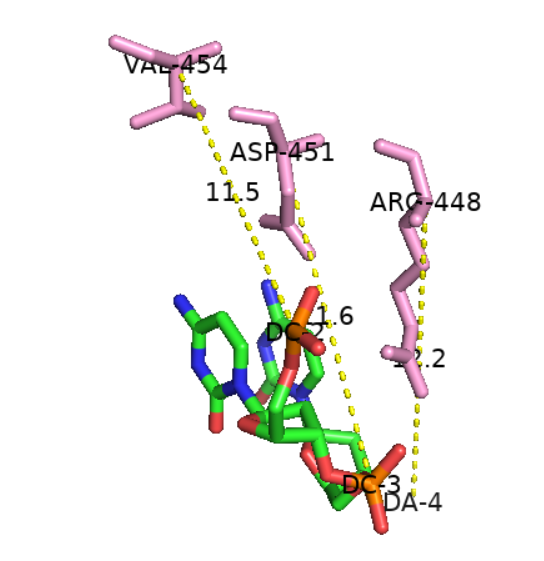
V454、D451、R448
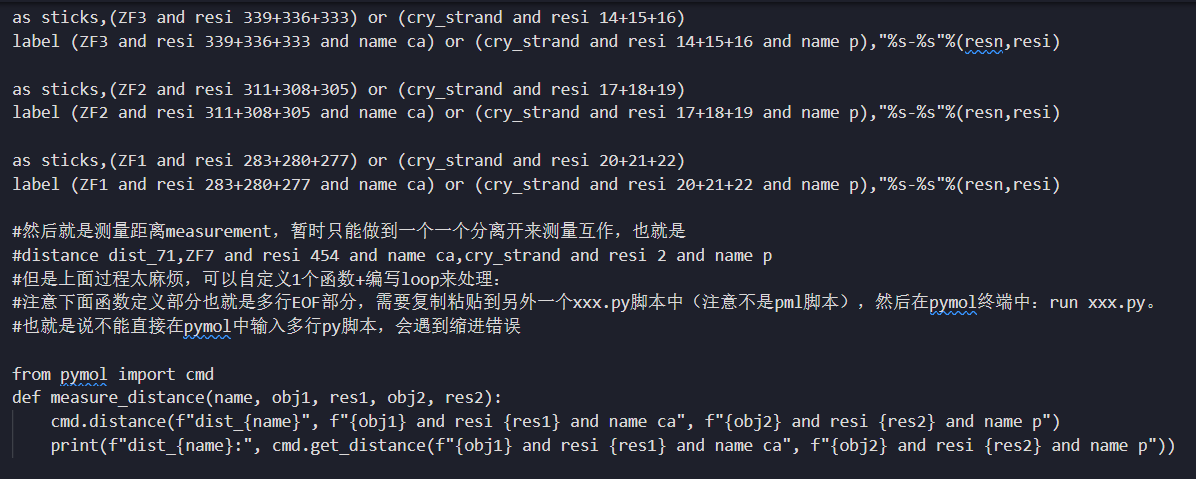
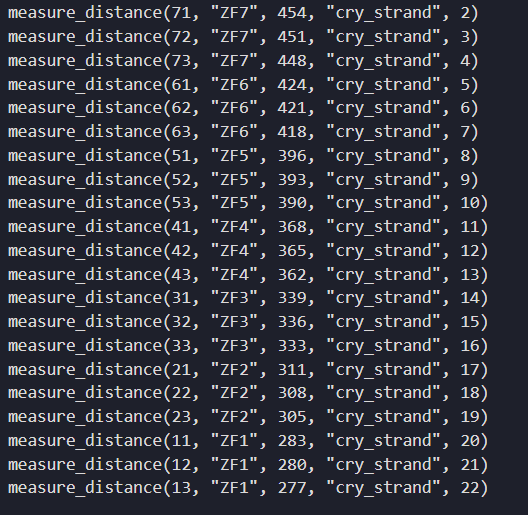
cry_strand
2C/3C/4A
所以其实只要ZF1_7以及DNA的结晶单链即可
所有对象都可以直接选择sele之后再as sticks再color
然后label的话其实是先选中DNA以及蛋白质对象,再使用
但是所有的原子都会显示
不选原子而是选择整个残基


(注意seq要对应上,别标错)
然后再是measurement

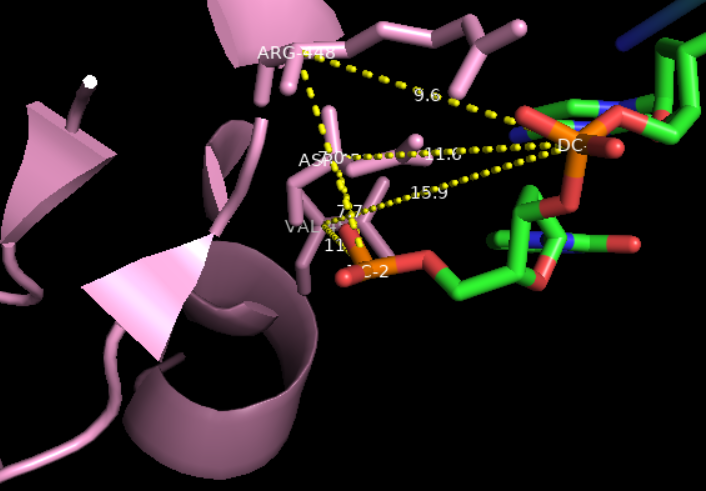
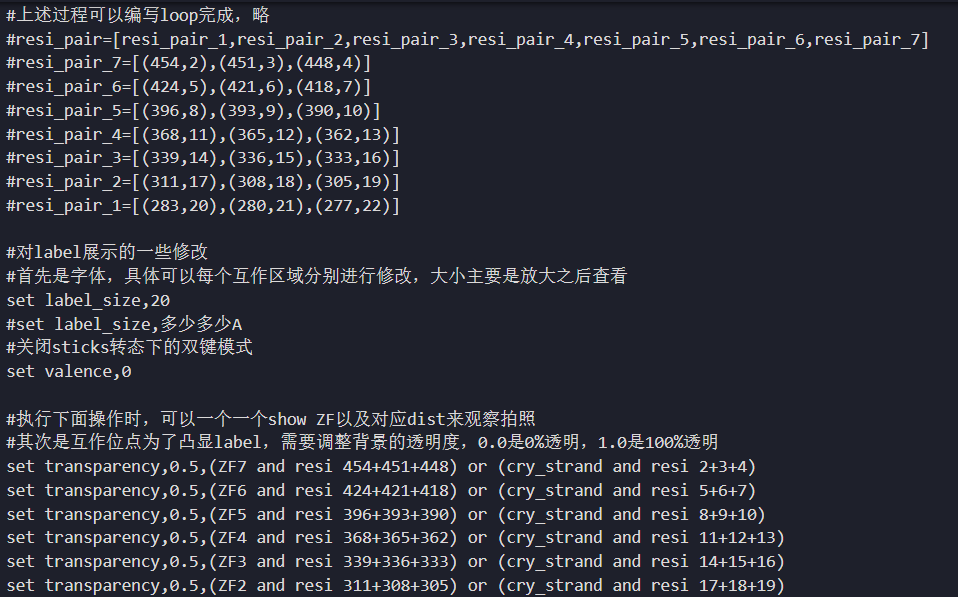
但是这样是3 vs 3的,如果要1vs1的话,只能使用1个残基1个残基的使用并标注
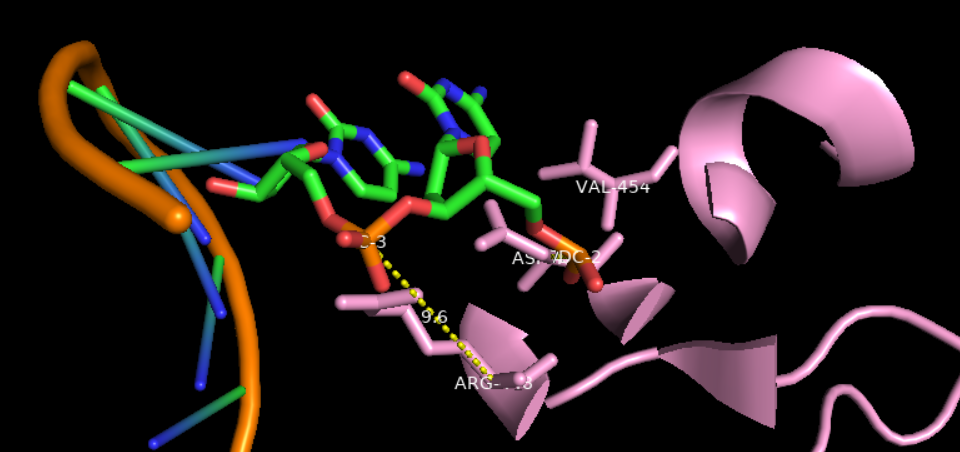
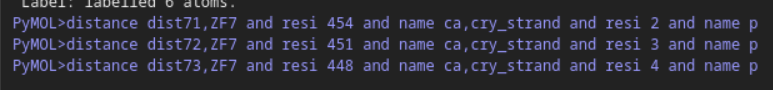

问题在于如何摆放:
还是靠不断移动,以显示数字效果
主要是残基背景的透明效果:
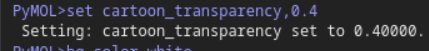
可以是选定残基之后的透明效果设置,也可以是是针对不同的show或者是as来设置对应的透明效果,
比如说上面是cartoon就是除了互作碱基之后的背景残基的透明效果,然后针对sticks的互作碱基的效果可以是设置为其他的透明效果;
当然作为背景的其他残基效果没必要更改,主要是修改在互作碱基之后的透明效果即可
也就是选中label的指令部分
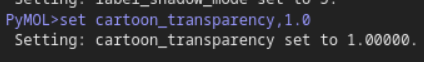
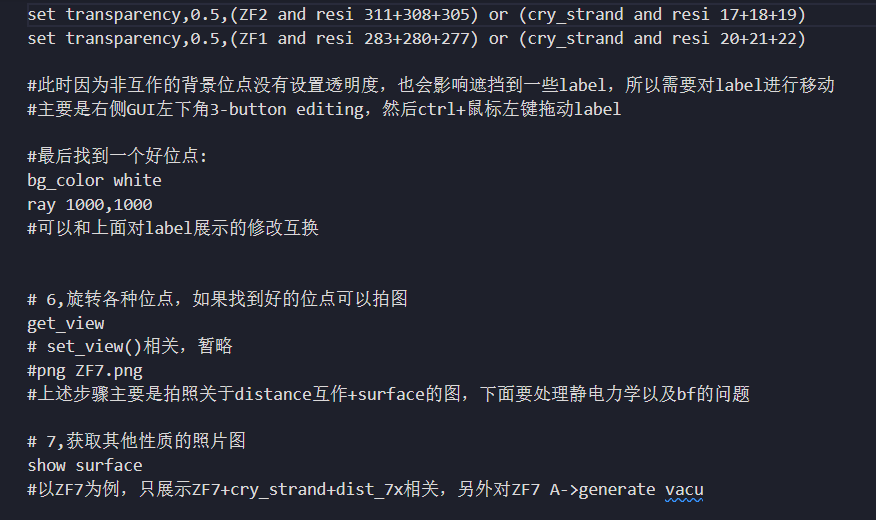
set transparency, 0.5, resi 454+451+448 and chain X and resi 1+2+3 and chain Y
但是效果基本没有,可能暂时无法针对集合修改?
只能先对show/as形式修改
应该是其余的背景设置最透明,然后互作的碱基略不透明,
比如说互作0.5透明,背景0.8
还是主要设置想要观察的互作残基的透明度,
其余的主要是移动label,如果在图上点击就是edit label

主要是:
再ctrl+鼠标左键移动
as和show的区别:
最好用show,无论是surface还是静电力学
show是显示,as是替换,如果有多种效果就使用show
然后在DNA以及蛋白质的相关选择中,and与&通用,以及选择的关键词是大小写不敏感的


上面ZF7处理完了其他的ZF1-6同样处理
ZF6:
M424、T421、Q418
G5/C6/A7
ZF5:
R396、K393、D390
G8/G9/G10/
ZF4:
R368、K365、E362
G11/G12/C13
ZF3:
R339、E336、T333
G14/C15/T16
ZF2:
N311、L308、T305
A17/G18/T19
ZF1:
R283、N280、R277
G20/A21/G22
(4)除了互作位点的拍照+distance的measurement,还可以做一些其他的操作:
比如说是静电力学的显示surface,或者是b-factor

还是放整体的:
DNA作为motif作为配体,在其中,然后显示整个ZF蛋白质的静电
草稿结束

















 2万+
2万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









