






经验上、文献上大量的蛋白质或者核酸比对工作是从一级序列开始的,这是基于一级序列决定二级结构,二级结构决定三级结构,而且一级序列有30%的相似性,那么两者的结构就具有较高的相似性这样的共识理论而来,这些理论也是基于现有的实验数据得来的,然而也存在很多例外的情况,如相似度高的蛋白序列,三级结构差异较大,或者相似度低的蛋白序列却具有较高的三级结构,又或者一些理论认为柔性蛋白可能具有更好的蛋白热稳定性,刚性太强反而在更剧烈的环境中更容易崩溃。这些反常识的理论反而更容易让人深思,如决定蛋白功能的到底是一级序列还是三级结构。
因此本章内容从三级结构比对开始摸索目标蛋白可能在某个环境中与那些蛋白可能有相同或者类似功能。
一、pdb文件下载
1)获取pdb文件
在pdb数据库https://www.rcsb.org/,搜索关键词或者序列,然后在左侧选择物种、精度、结构获取方法等可定制自己的需求。然后在右上侧selected处点击下载符号。
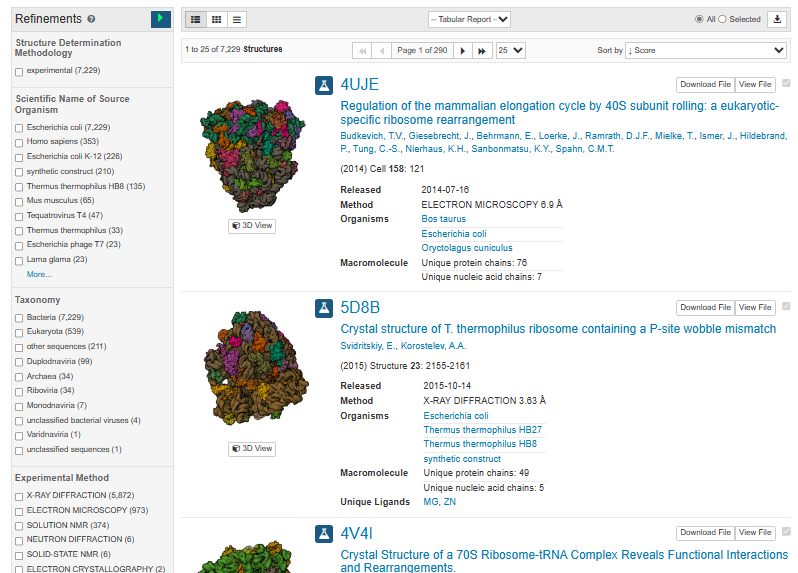
2)之后全部复制下面文本框中的pdb名,保存备用。
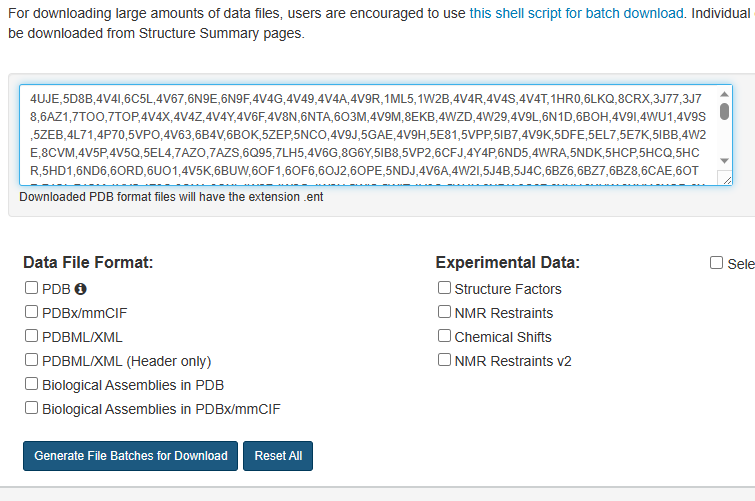
3)代码读取pdb并下载pdb文件:
import requests
from tqdm import tqdm
#读取pdb名称
f = open("/home/lxh/Documents/Lysin/E.coil_protein/Ecoil_protein","r")
for a in f.readlines():
a = a.split(",")
for b in tqdm(a):
request = requests.get("https://files.rcsb.org/download/" + b + ".pdb")
if request.status_code != 404:#判断有pdb文件才会下载,没有就跳过,防止报错
wget.download("https://files.rcsb.org/download/" + b + ".pdb","F:/biosoftware/gromacs/pymol/mutation/" + b + ".pdb")
二、处理pdb文件用于后续三维结构比对
import os
for c in os.walk("F:/biosoftware/gromacs/pymol/mutation"):
c = c[2]
#去水并多聚体变单聚体
for a in c:
if a[-3:] == "pdb":
f = open("F:/biosoftware/gromacs/pymol/mutation/" + a,"r")
f1 = open("F:/biosoftware/gromacs/pymol/structure_align/" + a,"a+")
for b in f.readlines():
f1.write(b)
if b.split()[0] == "TER":
break
f.close()
f1.close()
三、计算目标蛋白和其余蛋白的RMSD
注意此代码用于python中,后续生成的文件可以在pymol中运行
import os
for c in os.walk("F:/biosoftware/gromacs/pymol/structure_align"):
c = c[2]
f = open("F:/biosoftware/gromacs/pymol/三级结构比对.txt","w")
f.write("from pymol import cmd" + "\n" + "load F:/biosoftware/gromacs/pymol/GS4_CBD.pdb" + "\n" + 'output = open("E:/李小红资料/裂解酶治疗血流感染课题/protein/a.txt","w")' + "\n")
for a in c:
if a[-3:] == "pdb":
f.write("load F:/biosoftware/gromacs/pymol/structure_align/" + a + "\n" + 'output.write("' + a + '" + " " + str(cmd.align("GS4_CBD","' + a[0:-4] + '")[0]) + ''" "'' + str(cmd.align("GS4_CBD","' + a[0:-4] + '")[6]))' + "\n" + "delete " + a[0:-4] + "\n")
f.write("output.close()" + "\n")
f.close()四、筛选三维结构相似度高的PDB文件
将比对氨基酸较多(n),RMSD值较低(m)的值打印输出,然后为了保证结果可靠,可在pymol中肉眼观察下选中的几个pdb是否和目的蛋白三维结构具有较高的相似性。
#筛选相似度高的pdb
def similar(m,n):
f = open("E:/资料/血流感染课题/protein/a.txt","r")
for a in f.readlines():
if (float(a.split()[1]) < m) & (float(a.split()[2]) > n):
print(a)
f.close()将选中的三维结构具有高相似度的pdb在rcsb pdb数据库以及NCBI上查找其蛋白名称以及功能作用。大概推测目标蛋白可能具有选中蛋白的生物学功能。

















 7959
7959

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









