






虽然现在很多的生物公司都可以做BSA,还可以帮我们完成分析,但是其实我们给公司的很大一部分钱都是分析的钱,所以如果我们自己可以完成分析的话,可以大大节省经费,而且公司使用的参考基因组不一定是最新的,我们自己分析可以随意设置条件,使用最新的参考基因组。
1、win10如何下载linux子系统可以参考这个教程。
2、接着安装包的管理工具conda并替换源。
3、安装qtlseq,这里我们最好使用conda创造一个虚拟的环境,再安装qtlseq这个软件,否则可能会导致子系统环境被污染,或者某些依赖冲突报错。
conda creat -n bsa qtlseq #-n接的虚拟环境名字,后面是要安装的包4、下载测试数据,我的测试数据来自这篇文章“Identification of a cold-tolerant locus in rice(Oryza sativa L.) using bulked segregant analysis with a next-generation sequencing strategy”,根据文章编号SRR6327815, SRR6327816, SRR6327817, SRR6327818在ENA下载。参考基因组我用的是all.chrs.con。
5、运行下面的代码开始分析。
conda activate bsa #因为我是装在bsa这个环境中,所以我需要激活bsa才能使用qtlseq qtlseq -r all.chrs.fasta -p SRR6327815_1.fastq.gz,SRR6327815_2.fastq.gz --bulk1 SRR6327817_1.fastq.gz,SRR6327817_2.fastq.gz --bulk2 SRR6327818_1.fastq.gz,SRR6327818_2.fastq.gz --N-bulk1 21 --N-bulk2 21 -w 1000 -s 10 -T --species Rice -o out-n1 bulk1的混样数量 这里为SRR12412968样本 35个
-n2 bulk2的混样数量 这里为SRR12412969样本 37个
-t 调用的线程数
–mem 每个线程可调用的内存
-o 输出文件夹名字,必须原先不存在
-r 参考基因组序列位置
-p 其中一个亲本 一般用目标表型的亲本 这里用了突变体亲本
-T 启用trimmomatic质控
–trim-params trimmomatic的参数,如:[33,<ADAPTER_FASTA>:2:30:10,20,20,4:15,75].33为接头类型,后边是去除接头的参数
-q 是call SNP过程中要过滤的参数,最小的MQ值
-Q 是call SNP过程中要过滤的参数,最小的BQ值
-b1 bulk1的fastq或bam 文件
-b2 bulk2的fastq或bam 文件
-w 窗口的大小,默认是2000kb,单位是kb
-s 划窗大小,单位是kb
--species 可以选择RICE,小麦,玉米等
6、结果如下,可以看出与原文基本一致,在6号染色体。
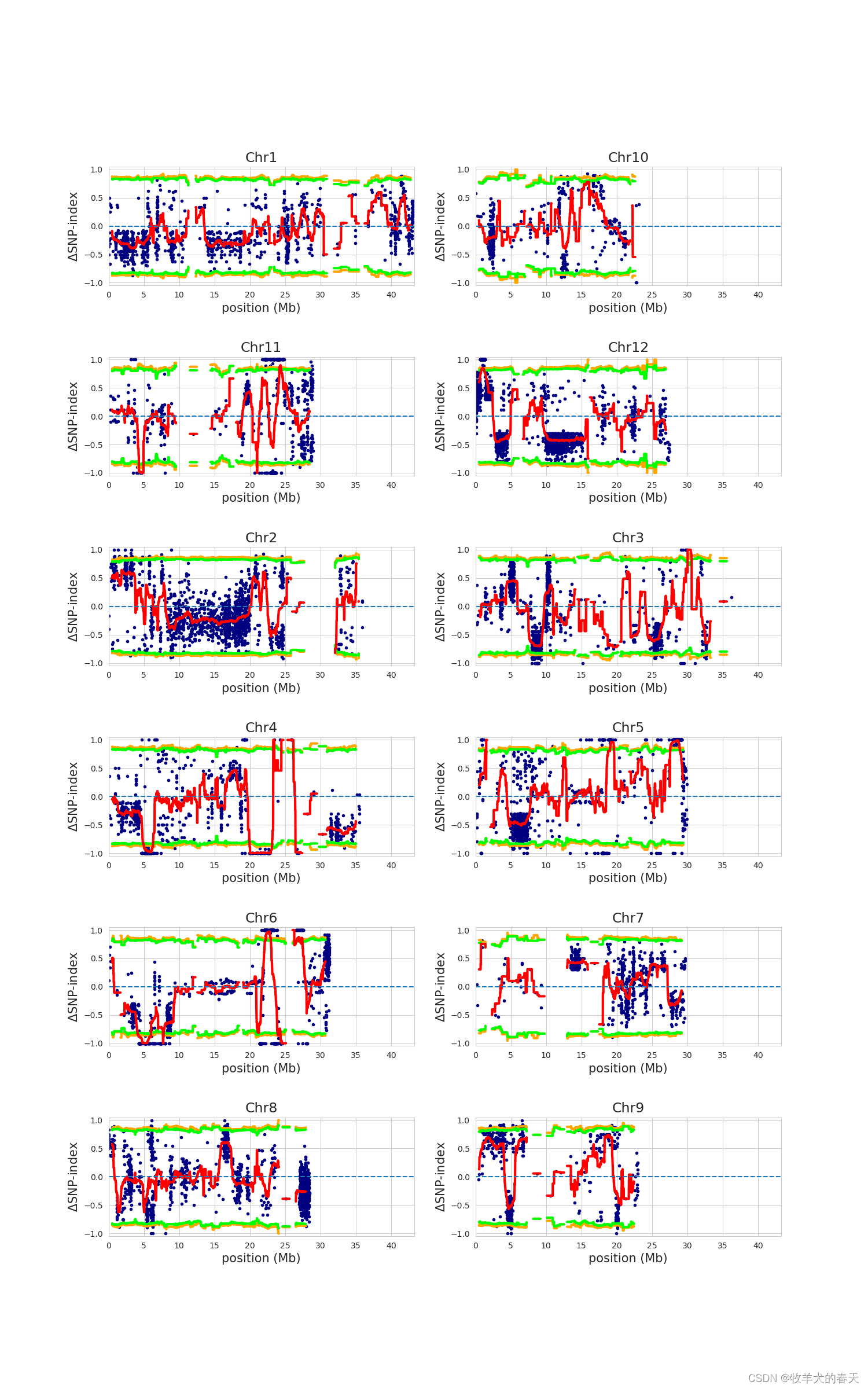
原文结果如下
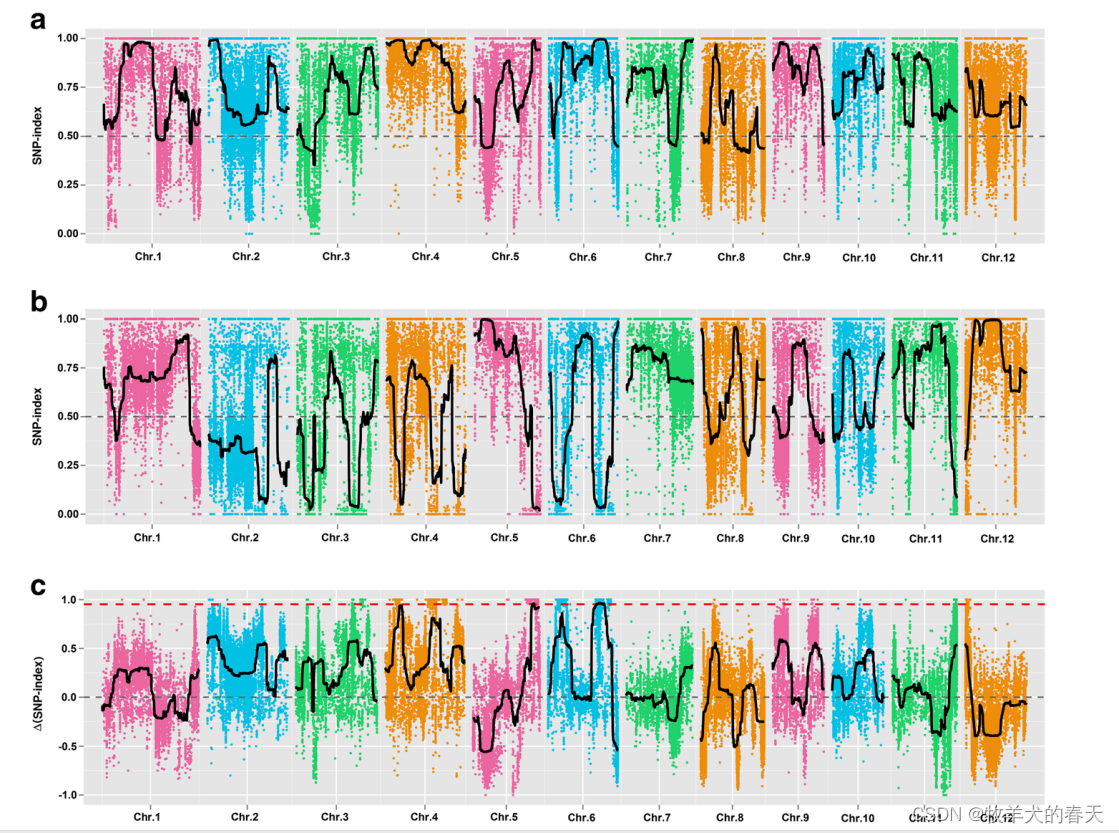
对mutmap感兴趣的也可以看看这篇,本文是对该博客的简化。















 7444
7444











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









